
Variant calling is the computational process by which a laboratory identifies variants in sequencing data. As one of the first steps in many NGS data analysis pipelines, accurate variant calling is often critical to downstream analysis and interpretation.
In turn, variant calling underpins many genomic applications, from helping to drive our understanding of genomics to facilitating the research into hereditary diseases and cancer genomics, paving the way to the future of precision medicine.
While differentiating between ‘true variants’ and ‘noise’ in sequencing data has presented an ongoing challenge in clinical research, advances in bioinformatics have fuelled increasingly accurate variant calling strategies. Here, we take a look at variant calling best practice through a modern lens.
Genomic variants – including single nucleotide variants (SNVs), small insertions and deletions (indels), and structural variants (SVs) – can be important markers of certain diseases. For instance, copy number variants (CNVs) – a type of SV - have been strongly associated with numerous cancers including solid tumours and haematologic malignancies. A more specific example is internal tandem duplication in FLT3, (FLT3-ITDs), a common SV in acute myeloid leukaemia (AML) often associated with poor prognosis and an aggressive form of the disease.
The original variant callers typically employed a single algorithmic approach2. In other words, one strategy was used to identify all the different types of genomic variants. Given the vast differences between variant types this is far from ideal.
Advances in bioinformatics have fuelled the development of variant calling software that combines multiple signatures and methods for the identification of different variant types (Figure 1).
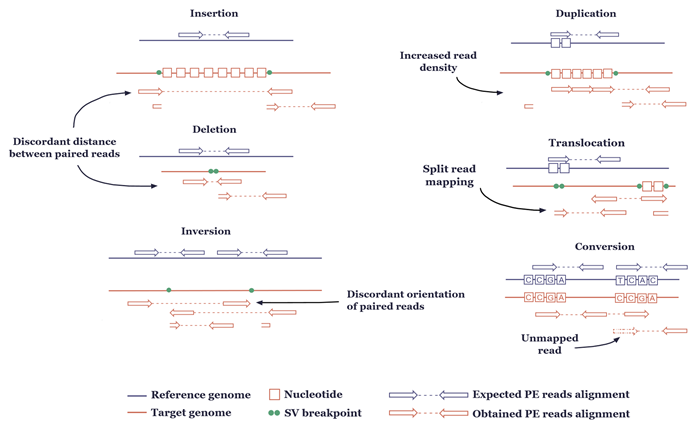 Figure 1: The major variant types and their characteristic read-alignment patterns for variant calling in NGS.
Figure 1: The major variant types and their characteristic read-alignment patterns for variant calling in NGS.
So, what software is best for your specific application?
SNVs are the most common type of mutations in human genomes and fortunately, they’re the most straight forward to identify. SNVs are a bit like typos – in that they are single base change in a nucleotide sequence. Typically, reads are aligned to a reference genome and, by employing a probabilistic methods or heuristics, the likelihood of each variation at each locus is predicted.
This type of analysis only works for germline mutations. Calling variants in somatic or cancer cells is more challenging. Tumour heterogeneity, the presence of both tumour and nontumor cells, and clonal expansions can generate many different variants at the same locus. In this scenario, it is advisable to run multiple samples from the same individual and use a variant calling software specifically designed for handling somatic variants.
Calling of SVs is even more challenging still. Because structural variants are 20 or more nucleotides in length, they are difficult to locate in individual short reads. But researchers have identified a number of patterns that can be used to identify SVs when mapping patient reads to reference genomes including (Figure 1)2:
Despite advances in bioinformatics, variant callers that identify multiple variant types could be described as a ‘jack of all trades, master of none’. To put it another way, there is no single tool which is optimal for all variant types and sequencing data. Therefore, if your study requires the accurate identification of multiple types of variants, it is worth considering the consolidation of variant call sets from multiple tools3.
You may have selected the best variant calling tool for your specific application but you can’t just feed raw sequencing reads into the software workflow. First, your NGS data needs to be adequately prepped to ensure optimal results. Typically, this involves:
When aligning your reads, it is important to be selective about your choice of reference genome. A poor choice can adversely affect the quality and validity of your results. Additionally, should your study call for it, it is advisable to include alternative haplotypes of hypervariable regions, such as the major histocompatibility locus (MHC)2.
During alignment, there is a balance to be struck between sensitivity and specificity. If sensitivity is increased this may lead to a lack of specificity. When initially aligning your reads to the reference genome, it is preferable to prioritize sensitivity over specificity to ensure that variants are not overlooked.
Some software developers recommend additional processing steps. For example, the GATK Best Practices workflow suggests performing base quality score recalibration (BQSR) as well as local realignment around indels4. These steps require a high amount of computing power and may only result in marginal improvements to variant calls – so the choice is yours to make.
Although variant calling itself is a bioinformatics process, the quality of its output is largely dictated way before the sequencing data is even generated. As the computer science adage goes, ‘garbage in, garbage out’.
Throughout your workflow, from collection and storage to extraction method and sequencing strategy, there are various factors that can affect your ability to accurately identify variants.
For example, in clinical research, formalin-fixed paraffin-embedded (FFPE) samples are often used to archive specimens. While a useful method for preserving tissue blocks, FFPE samples often yield low quantities of degraded DNA, containing formalin-induced mutations. Not only can this result in sub-optimal sequencing data but it can also make it difficult to distinguish between true and damage-induced low-frequency mutations.
While processing multiple samples per individual can increase the specificity of variant calling, these samples are often precious and sequencing can quickly become expensive. The use of repair enzymes - such as the SureSeq™ FFPE DNA Repair Mix – on DNA extracted from FFPE samples can help remove a broad range of damage and in turn, help increase your confidence in variant calls.
Both choice in extraction method and sequencing platform can have important consequences on variant calling. For example, short reads are popular and cost-effective strategy for SNV detection but read length may be too short to effectively identify larger variations. By contrast, long read sequencing can span over hundreds of kilobases and therefore large repeats and regions – which can assist the calling of larger SVs2.
Finally, different sequencing strategies offer different depth and breadth of sequencing coverage that can influence variant calling results. For instance, only whole genome sequencing (WGS) allows simultaneous and comprehensive detection of all variant types. However, panel and exome sequencing may enable more sensitive detection of SNVs at low allele frequencies, such as in cancer germline variants, due to their higher sequencing depths3.
Multi-gene panels are an accurate and cost-effective method for evaluating a subset of genes associated with a clinical presentation or disease. For instance, the SureSeq CLL + CNV Panel from OGT provides comprehensive coverage of 13 key genes and 5 chromosomal regions implicated in CLL progression. By simultaneously detecting SNVs and indels, as well as exon-level to whole gene CNVs, sequencing panels such as those offered by OGT can alleviate the burden of running multiple assays and streamlines your CLL research to deliver a comprehensive genomic profile for each CLL sample using a single workflow.
Discover OGT’s range of SureSeq NGS Panels
The choice of variant calling software is so vast it can be overwhelming. Moreover, integrating variant calling into a comprehensive NGS workflow takes time, expertise, and resources. Automated data analysis of sequencing data is becoming increasingly important; especially in clinical research where time and resources may be stretched.
Some providers are addressing this challenge by designing comprehensive, intuitive and in some instances, automated software that streamlines the analysis workflow. OGT’s complimentary Interpret NGS Analysis Software, for example, provides automated data analysis based on predefined settings minimising the need for user intervention while maximising the consistency and speed of data analysis.
Being able to reliably call a range of variant types can take significant optimisation and analysis time for most laboratories. Highly optimised software such as Interpret simplifies process and delivers accurate calling of SNVs and indels, as well as structural aberrations, including ITDs, PTDs, CNVs, LOH and translocations. To make NGS data analysis even more effortless, OGT can provide individualised options through its plug-in infrastructure, allowing Interpret software to be tailored to your laboratory’s specific requirements.
Discover how OGT’s Interpret NGS Analysis Software can help you optimise your workflow!
NGS has transformed our understanding of human health and disease by enabling large-scale genomic sequencing efforts. In turn, sequencing technologies have been leveraged as frontline tools for inherited diseases, advancing the possibilities of precision medicine, and driven deeper insights into the connection between genome and disease.
Accurate variant calling is the backbone of many of these genomic studies and translational applications. Increasingly improved algorithms and optimized computing will continue to improve variant calling procedures. However, implementing best practices – such as optimal storage and preparation of samples and rigorous pre-processing of sequencing data – will remain central to variant calling well into the future.

A high-quality sequencing library is the linchpin to generating good sequencing data. We discuss our six top tips to help you improve your sequencing library.
Read
We discuss the development and current state of sequencing technologies, and where the future of NGS may take us...
Read
FISH is a cytogenetic technique utilised in labs to detect chromosomal abnormalities in both cancer and constitutional specimens. In this blog learn about the advantages of FISH...
Read
This blog will discuss FLT3’s normal function, its implications in myeloid malignancies, and the role of NGS in genetic identification and disease management of patients with FLT3 genetic alterations.
Read
While liquid biopsy may present an attractive alternative to a solid biopsy, it also has limitations. Here, we shed light on some advantages, limitations, and future outlook for liquid biopsy in oncology clinical practice.
Read
Find out about the benefits and differentiating factors of the three most commonly used NGS technologies; targeted gene panels, whole-exome sequencing (WES) and whole-genome sequencing (WGS).
Read
Next generation sequencing (NGS) is now in routine use for a broad range of research and clinical applications. Facilitating the detection of a wide variety of mutations, focus has never been higher on the value of making the correct choice for the initial sequence enrichment step, which, if poorly designed, can be a source of bias and error in the downstream sequencing assay.
Read Visit USA site
Visit USA site Visit Canada site
Visit Canada site
