
Next generation sequencing (NGS) is now in routine use for a broad range of research and clinical applications. Facilitating the detection of a wide variety of mutations, focus has never been higher on the value of making the correct choice for the initial sequence enrichment step, which, if poorly designed, can be a source of bias and error in the downstream sequencing assay.
The two main enrichment strategies commonly used are amplicon (PCR)-based and hybridisation-based. Each has its own advantages and disadvantages. Amplicon has the advantage of offering a simpler workflow, it is faster, the ongoing cost of running an amplicon-based technology is relatively low and can accommodate a lower sample input. Hybridisation, on the other hand, is a more robust technique and has the advantage of providing better uniformity of coverage, fewer false positives, and superior variant detection due to fewer PCR cycles.
An important limitation of amplicon-based approaches is the difficulty with assessing the true complexity of the library – every molecule that you sequence will start and end with the same base (as determined by PCR primers used for Multiplex PCR) so it’s virtually impossible to remove duplicates without using molecular identifiers, which whilst excellent for deduplication and error correction, can reveal high levels of duplication and low complexity. This over representation of some regions, if those regions contain mutations, either true or PCR induced errors, has the potential to impact downstream results.
Another disadvantage is fairly poor uniformity of coverage – certain regions of the genome will amplify better than others and therefore better depth of sequencing will be achieved in those regions, figure 1.
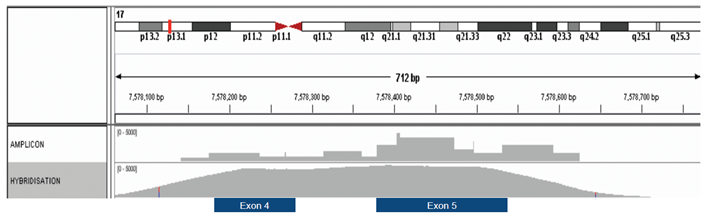 Figure 1. Hybridisation-based enrichment delivers more uniform coverage. Comparison of amplicon and hybridisation-based enrichment of exons 4 and 5 of the TP53 gene.
Figure 1. Hybridisation-based enrichment delivers more uniform coverage. Comparison of amplicon and hybridisation-based enrichment of exons 4 and 5 of the TP53 gene.
Random fragmentation of DNA, the initial step in hybridisation-based approaches, means that all sequenced fragments are unique, starting and ending with a different base. This allows removal of PCR bias introduced during the library prep stage and this is critical in determining the true complexity of the starting material. The approach is also very tolerant when it comes to fragmented DNA, with even short fragments being captured relatively well.
With only limited quantities of starting material often available, it is important to note that the actual number of templates available is relatively low, so duplication rates can increase significantly. With hybridisation assays, these can be removed computationally to leave clean, high-quality data. With amplicon assays, this is not possible without the use of molecular barcodes, and the resulting data may be skewed by the over-amplification of a small number of fragments.
See how researchers from the BC Cancer Agency in Vancouver, British Columbia, evaluated both hybrid capture and amplicon-based targeting in developing their analysis pipeline to study lymphocytic malignancies: https://jmd.amjpathol.org/article/S1525-1578(17)30425-7/fulltext
As one of the first steps in many NGS data analysis pipelines, accurate variant calling is often critical to downstream analysis and interpretation. Here, we take a look at variant calling best practice through a modern lens.
Read
A high-quality sequencing library is the linchpin to generating good sequencing data. We discuss our six top tips to help you improve your sequencing library.
Read
We discuss the development and current state of sequencing technologies, and where the future of NGS may take us...
Read
FISH is a cytogenetic technique utilised in labs to detect chromosomal abnormalities in both cancer and constitutional specimens. In this blog learn about the advantages of FISH...
Read
This blog will discuss FLT3’s normal function, its implications in myeloid malignancies, and the role of NGS in genetic identification and disease management of patients with FLT3 genetic alterations.
Read
While liquid biopsy may present an attractive alternative to a solid biopsy, it also has limitations. Here, we shed light on some advantages, limitations, and future outlook for liquid biopsy in oncology clinical practice.
Read
Find out about the benefits and differentiating factors of the three most commonly used NGS technologies; targeted gene panels, whole-exome sequencing (WES) and whole-genome sequencing (WGS).
Read Visit USA site
Visit USA site Visit Canada site
Visit Canada site
