
Garbage in, garbage out is an expression commonly used in science to express that poor quality DNA libraries will generate erroneous sequencing results. So, when it comes to next-generation sequencing (NGS) how do you ensure that what you put in is good enough to get the quality of results that you need?
A high-quality sequencing library is the linchpin to generating good sequencing data. Ideal libraries consist of purified target sequences of the right size distribution, ligated to appropriate sequencing adaptors, and in sufficient concentrations for your chosen sequencing platform.
While getting here may take some in-house optimisation – dependent on your specific sample type, application and chosen sequencing platform – all library preparation workflows can benefit from some universal tips and tricks. In this blog, we discuss our six top tips to help you improve your sequencing library.
Whether you’re identifying sequence-based biomarkers in haematological malignancies or surveying gene expression changes in solid tumours, the first common step to nearly all NGS studies is the extraction of nucleic acids. Choosing a sub-optimal extraction method can ultimately compromise the quality of your library. For example, inadequate cell lysis could result in insufficient yields while carried over contaminants can be detrimental to enzymes used in downstream steps such as ligation.
An incomplete extraction could also introduce bias into your dataset. A successful gene expression analysis of multiple cell types, for example, is contingent on the ability to access mRNA in the same proportion in which they exist in the body. Consequently, cell lysis must account for any variation in the severity needed to lyse different cell types.
Some samples prove particularly recalcitrant to DNA extraction. Formalin-fixation, paraffin-embedded (FFPE) samples, the most frequently used method of tissue preservation, are often challenging to nucleic acid extraction.
The major challenge of nucleic acid extraction from FFPE tissue is the prevalence of chemical crosslinking that binds nucleic acids to other proteins and DNA or RNA strands. This can result in impure, degraded and fragmented nucleic acid samples that are sub-optimal for sequencing libraries.
Damaged DNA not only means lost information but can also lead to false conclusions. For example, extensive DNA damage may result in difficulty distinguishing between true and damaged-induced low frequency mutations. However, there are steps that can be taken to decouple these cross links, preserving original complexity and delivering high-quality sequencing data. For example, SureSeq™ FFPE DNA Repair Mix is a mixture of enzymes that has been optimised to remove a broad range of damage that can cause artefacts in sequencing data.
PCR cycles can be necessary to generate enough material for sequencing. However, they also increase the risk of introducing bias into your sequencing library. AT-rich regions of DNA, for example, are generally amplified more efficiently than GC-rich regions as it’s easier to denature the double-stranded DNA.
Amplification is increasingly essential for low concentration samples. However, with decreasing nucleic acid concentrations and increasing PCR cycles comes an increasing risk of an erroneous dataset. When input amounts are low, bias amplification of a small subset of nucleic acid sequences can cause a significant drop in diversity and a large skew in your dataset compared to when input amounts are high.
So, what’s the answer?
Simply reducing the number of PCR cycles isn’t always possible when there’s a threshold concentration needed for sequencing. The first step would be to increase the amount of starting material and optimise your extraction steps. But, again, increasing starting material is often not an option.
Therefore, it is also important to choose the optimal sequencing library kit. If amplification is necessary, select a kit that offers high-efficiency end repair, 3’ end ‘A’ tailing and adaptor ligation as this can help minimise the number of required PCR cycles.
Find out how OGT’s Universal NGS Complete Workflow solution gives exceptionally low levels of duplication.
Additionally, selecting a hybridisation enrichment strategy rather than an amplicon enrichment approach is preferable - minimizing errors is critical to experimental outcome such as for variant detection. Although amplicon approaches offer a simpler and faster workflow, hybridisation is a much more robust technique that yields better uniformity of coverage, fewer false positives, and superior variant detection due to the requirement of fewer PCR cycles.
Read more about hybridisation vs. amplicon
Unique molecular identifiers (UMIs) and Unique dual indexes (UDIs) are strategies used to allow multiplexing and enable accurate demultiplexing during downstream data analysis.
UMIs are short molecular sequences that work like molecular barcodes. By uniquely tagging each molecule in a sample library with a UMI, true variants can be differentiated from errors introduced during library preparation, target enrichment, or sequencing. This approach can be particularly useful if you are trying to identify low frequency variants and to efficiently identify any PCR errors.
Indexing using UDIs involves the ligation of two different index barcodes (i5 and i7) to every sequence molecule. However, while UMIs are tagged to each molecule in a sample, indexes are assigned to a specific library. In combinatorial indexing, each combination of indexes is different however, each i7 and i5 is used multiple times on a plate of libraries (Figure 1). Non-redundant indexing uses UDIs where each library has a completely unique i7 and i5 (Figure 1). This strategy has been incorporated into more modern workflows – such as the use of combinatorial indexing to prepare sequencing libraries – as it allows more accurate demultiplexing and avoids index hopping.
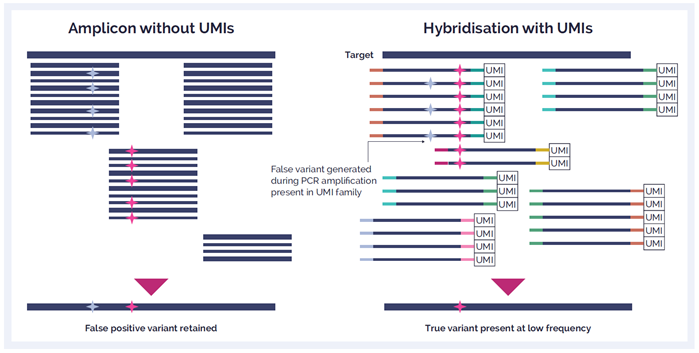 Figure 1 (above). Combinatorial indexing uses a plate matrix approach leading to unique index sets of non-unique indexes. Non-redundant indexing uses completely unique indexes across the whole plate.
Figure 1 (above). Combinatorial indexing uses a plate matrix approach leading to unique index sets of non-unique indexes. Non-redundant indexing uses completely unique indexes across the whole plate.
Library preparation can require a significant amount of hands-on time in the lab. Not only can this be laborious, but the more steps in your protocol and the longer you spend manipulating your samples, the more chance you have to make a mistake.
No matter how experienced or engaged you are, we all make mistakes. In library preparation, human error can lead to cross-contamination or the introduction of contaminants – including nucleic acids or components that reduce the effectiveness of sequencing. Overall, this may reflect in your results quality or even generate incorrect data.
Some suppliers have optimised their kits to streamline the standard library preparation protocol. For instance, OGT has extensively tested enzymes and buffers to minimise the number of hands-on steps and overall processing time needed for its Universal NGS Complete Workflow. By combining the multi-enzymatic fragmentation and end repair and A-tailing step you’ll need to carry out fewer clean-up and QC steps which helps to minimise human errors.
But if you really want to reduce the risk of human error then it’s time to consider automation. Automation can partially or entirely remove the need for any hands-on time. OGT’s Universal NGS Library Complete Workflow, for example, has been designed to remove the need for secondary equipment such as ultrasonicators and vacuum concentrators easing the development of a more streamlined automation workflow. Indeed, OGT have developed a tailored automation workflow that not only improves throughput but reduces contamination risk and can improve the reliability and quality of your sequencing library.
After meticulously preparing your sequencing library, you don’t want to fail at the final hurdle! Quantifying and normalising your library are the final steps before you begin your sequencing run – and it’s important to make sure this is accurate.
Overestimating your library concentration will result in loading the sequencer with too little input and in turn, reduced coverage. On the other hand, if you underestimate your library concentration, you can overload the sequencer and reduce its performance. Additionally, if you are pooling libraries then incorrect quantification can lead to uneven library concentrations and ultimately, inconsistencies in data quality.
There are numerous methods for nucleic acid quantification including; UV absorption, intercalating dyes, quantitative PCR (qPCR) and droplet digital emulsion PCR. While there is no unambiguous gold standard, fluorometric methods risk overestimating the library concentration because it measures all double stranded DNA in the sample. Conversely, qPCR methods are extremely sensitive and only measure adaptor ligated-sequences.
Samples are often precious – maybe they’re irreplaceable patient samples or you’ve spent months collecting samples from difficult-to-culture cells. Now, imagine your frustration when your library preparation fails because an enzyme mix was stored at the wrong temperature!
While it may sound obvious, following laboratory best practices is essential to obtaining a good sequencing library:
For more expert tips on preparing samples for NGS, visit our NGS Success page.
To get the most out of your NGS experiment, it is important to consider the factors that determine the quality and quantity of your final library. While some of the challenges to creating a high-quality library are sample- or application-specific, following these tips and selecting the optimal library preparation kit are sure to help you generate reliable sequencing data.
From universal library preparation to methods specific to solid tumour samples, OGT’s NGS library preparation kits are optimised for your application. Working hand-in-hand with both SureSeq targeted cancer enrichment panels, as well as with the CytoSure® Constitutional NGS kit you’ll get the most sensitive and reproducible variant detection with excellent coverage uniformity. Discover a wide range of SureSeq and CytoSure NGS products designed to support you across your entire NGS workflow.
As one of the first steps in many NGS data analysis pipelines, accurate variant calling is often critical to downstream analysis and interpretation. Here, we take a look at variant calling best practice through a modern lens.
Read
We discuss the development and current state of sequencing technologies, and where the future of NGS may take us...
Read
FISH is a cytogenetic technique utilised in labs to detect chromosomal abnormalities in both cancer and constitutional specimens. In this blog learn about the advantages of FISH...
Read
This blog will discuss FLT3’s normal function, its implications in myeloid malignancies, and the role of NGS in genetic identification and disease management of patients with FLT3 genetic alterations.
Read
While liquid biopsy may present an attractive alternative to a solid biopsy, it also has limitations. Here, we shed light on some advantages, limitations, and future outlook for liquid biopsy in oncology clinical practice.
Read
Find out about the benefits and differentiating factors of the three most commonly used NGS technologies; targeted gene panels, whole-exome sequencing (WES) and whole-genome sequencing (WGS).
Read
Next generation sequencing (NGS) is now in routine use for a broad range of research and clinical applications. Facilitating the detection of a wide variety of mutations, focus has never been higher on the value of making the correct choice for the initial sequence enrichment step, which, if poorly designed, can be a source of bias and error in the downstream sequencing assay.
Read Visit USA site
Visit USA site Visit Canada site
Visit Canada site
