
Want to download this as a PDF? Download now
![]() With advances in next-generation sequencing (NGS) technology, genetic information can be extracted from an increasingly diverse range of samples. Formalin-fixed, paraffin-embedded (FFPE) storage is a standard method for archiving tissue biopsies and these biopsies can be used in NGS, for example to study cancer development and progression.
With advances in next-generation sequencing (NGS) technology, genetic information can be extracted from an increasingly diverse range of samples. Formalin-fixed, paraffin-embedded (FFPE) storage is a standard method for archiving tissue biopsies and these biopsies can be used in NGS, for example to study cancer development and progression.
However, the quality of DNA in FFPE samples is often severely damaged and compromised compared to other methods of sample storage. Consequently, it may be difficult to distinguish between true low frequency mutations and damage-induced low frequency false positives. In these samples, hybridisation-based target enrichment (where input DNA has been sheared into short, fragments and selectively captured) provides superior performance compared to amplicon-based enrichment. This is due to the superior tolerance of the hybridisation approach for the fragmented DNA routinely found in FFPE samples. Hybridisation-based enrichment also provides greater uniformity of coverage, fewer false positives and superior variant detection due to use of fewer PCR cycles1.
To improve the sequencing results of FFPE samples even further, a DNA pre-treatment step can be introduced to address different types of DNA damage. OGT’s SureSeq™ FFPE DNA Repair Mix uses a mixture of enzymes to repair a range of DNA defects including deamination of cytosine, nicks and gaps, oxidised bases, and blocked 3’ ends.
In this study, carried out in collaboration with Horizon Discovery, formalin-compromised DNA (fcDNA) samples of differing severity were repaired with the SureSeq FFPE repair mix and sequenced using a SureSeq custom hybridisation-based panel. The aim of the study was to investigate the effect of the repair mix and also the performance of the hybridisation-based enrichment method on DNA with varying levels of damage by measuring critical parameters at different points in the sequencing protocol:
When studying FFPE tumour biopsies, low DNA starting material is also a frequently encountered issue. In order to study the effect of this, tests were carried out with varying amounts of DNA starting material to determine sequencing sensitivity when DNA availability is limited.
To investigate the effect of the SureSeq FFPE DNA Repair Mix, Horizon Discovery provided three fcDNA samples with ‘mild’, ‘moderate’ and ‘severe’ damage2 . The samples were investigated in duplicate before and after repair; the amounts of input DNA were 200, 100, 50 and 10 ng (Figure 1). All samples were sheared using a Covaris S220 focused-ultrasonicator and prepared using the SureSeq NGS Library Preparation Kit (cat. no. 500070).
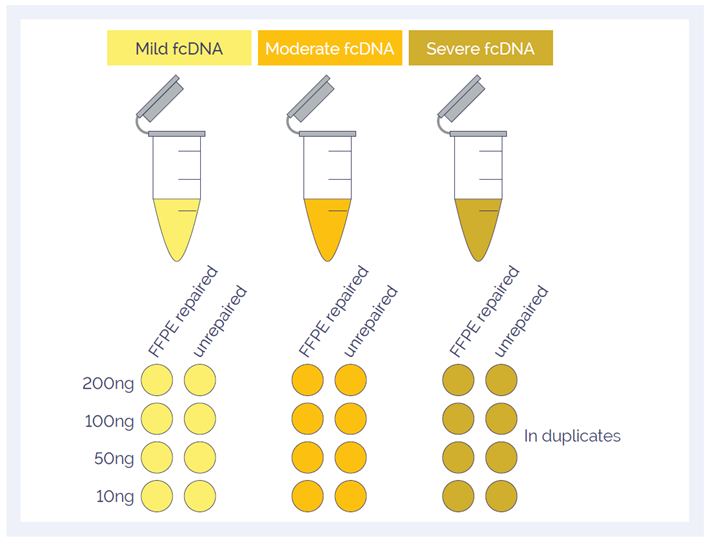 Figure 1: A total of 48 samples were investigated to study the effect of DNA quality, input amount and DNA repair.
Figure 1: A total of 48 samples were investigated to study the effect of DNA quality, input amount and DNA repair.
Enrichment by hybridisation was completed with an 8.7 Kb custom hot-spot panel designed to target the variants listed in Table 1. The subsequent post-capture libraries were sequenced on an Illumina MiSeq® using a v2 300 cycles kit (cat. no. MS-102-2002). 16 samples were run on a MiSeq lane.
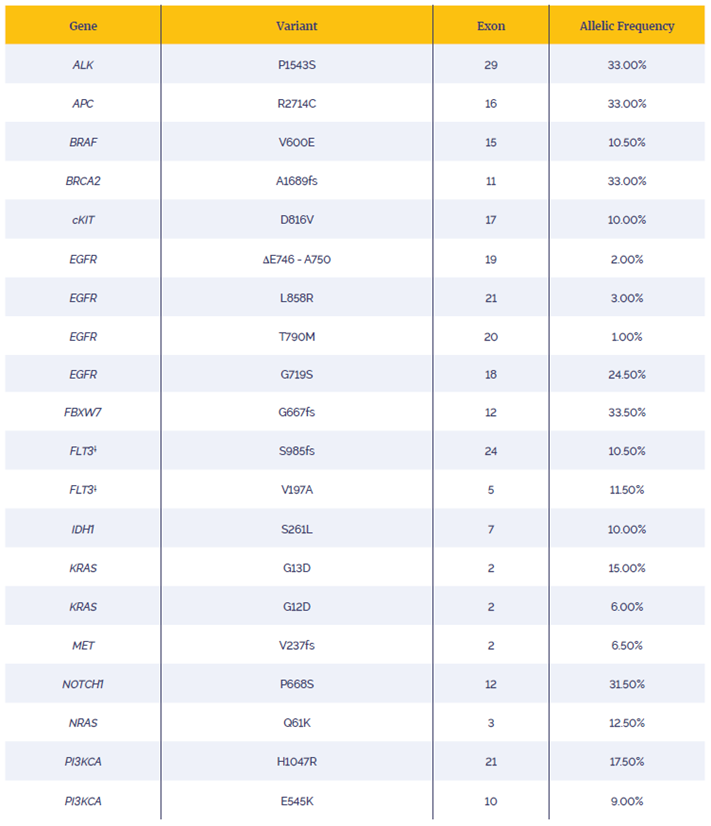 Table 1: List of variants examined. All mutations verified by ddPCR except**.
Table 1: List of variants examined. All mutations verified by ddPCR except**.
To confirm the quality of the three samples, DNA integrity numbers (DIN), an indication of DNA quality were determined using the Agilent TapeStation. Both DIN and DNA length distribution confirmed levels of DNA damage (Figure 2).
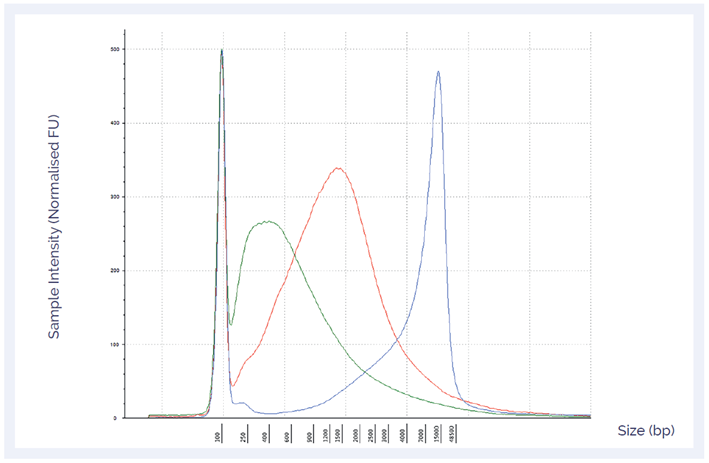 Figure 2: DNA length distribution and DIN can give an indication of the level of DNA damage in a sample. The sample with mild damage had the highest DIN (6.6) (blue), followed by the moderate (3.2) (red) and severe (1.9) (green) samples.
Figure 2: DNA length distribution and DIN can give an indication of the level of DNA damage in a sample. The sample with mild damage had the highest DIN (6.6) (blue), followed by the moderate (3.2) (red) and severe (1.9) (green) samples.
Higher library yields improve the complexity of the library, which in turn leads to better quality sequencing data. Figure 3 shows the effect of DNA repair on pre-capture library yields in the most severely damaged sample with different quantities of input DNA; the mild and moderate samples showed similar results. The difference in peak height at around 200 base pairs demonstrates that more DNA was available for capture and sequencing when the FFPE repair mix was used.
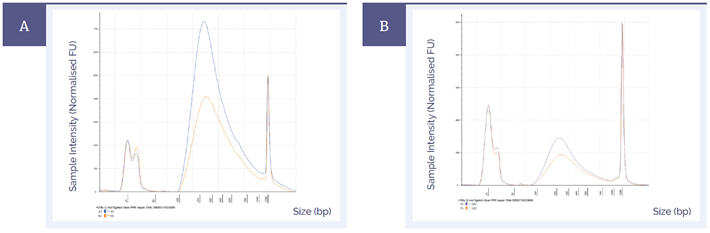 Figure 3: Pre-capture library yields of fcDNA with ‘severe’ damage treated with SureSeq FFPE DNA Repair Mix (blue) and without treatment (orange). The amounts of input DNA were 200 ng [A] and 50 ng [B].
Figure 3: Pre-capture library yields of fcDNA with ‘severe’ damage treated with SureSeq FFPE DNA Repair Mix (blue) and without treatment (orange). The amounts of input DNA were 200 ng [A] and 50 ng [B].
The increase in library yield had a positive effect on target coverage in all tested samples (Figures 4 and 5). Improvements in mean target coverage (MTC) varied from 20% up to 50%, showing the positive effect of higher library yields.
DNA treated with the FFPE repair mix also demonstrates a strong sequencing performance at low input amounts, maintaining a MTC (after removal of PCR duplication) of over 1000x for all samples at 100 ng input and over 500x at 50 ng input. This result shows that even when there are only small amounts of DNA available for analysis, it may still be possible to achieve a high level of coverage when using the FFPE repair mix. The result in Figure 5 also shows the direct link between both the quality and the quantity of DNA on variant calling accuracy.
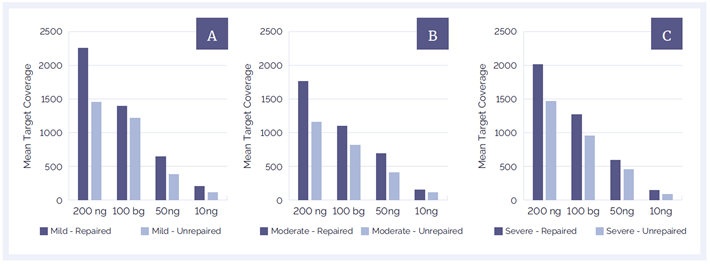 Figure 4: DNA repair significantly improves MTC and helps to improve coverage at all amounts of starting material and all levels of DNA damage; [a]: mild damage, [b]: moderate damage, [c]: severe damage.
Figure 4: DNA repair significantly improves MTC and helps to improve coverage at all amounts of starting material and all levels of DNA damage; [a]: mild damage, [b]: moderate damage, [c]: severe damage.
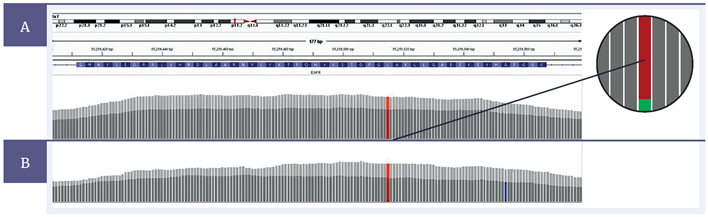 Figure 5: Comparison of the coverage in the region of an EGFR L858R mutation, 3% variant allele frequency (VAF), between a severely FFPE compromised sample treated with the SureSeq FFPE Repair Mix (light grey) and one without treatment (dark grey). At 200 ng input, [A] , following treatment, the total number of reads increases from 2707 to 3860, with the number of supporting reads for the mutation (see expanded illustration) increasing from 85 to 112 (after removal of PCR duplication). At 10 ng input, [B] , following treatment, the total number of reads increases from 216 to 341, with the number of supporting reads for the mutation increasing from 5 to 13. Note that the combination of low (10 ng) input and poor DNA quality increases the likelihood of the generation of false positive mutations (blue) and other spurious data. Total read counts are after the removal of PCR duplication.
Figure 5: Comparison of the coverage in the region of an EGFR L858R mutation, 3% variant allele frequency (VAF), between a severely FFPE compromised sample treated with the SureSeq FFPE Repair Mix (light grey) and one without treatment (dark grey). At 200 ng input, [A] , following treatment, the total number of reads increases from 2707 to 3860, with the number of supporting reads for the mutation (see expanded illustration) increasing from 85 to 112 (after removal of PCR duplication). At 10 ng input, [B] , following treatment, the total number of reads increases from 216 to 341, with the number of supporting reads for the mutation increasing from 5 to 13. Note that the combination of low (10 ng) input and poor DNA quality increases the likelihood of the generation of false positive mutations (blue) and other spurious data. Total read counts are after the removal of PCR duplication.
In order to assess the effect of increased MTC on the ability to call variants with greater accuracy the concordance between the NGS results of the fcDNA samples and Horizon Discovery’s Reference Standards was investigated using OGT’s SureSeq Interpret software. Table 2 details the 20 variants analysed: 15 single nucleotide variants (SNVs) and 5 deletions, with VAF varying between 1% and 33%.
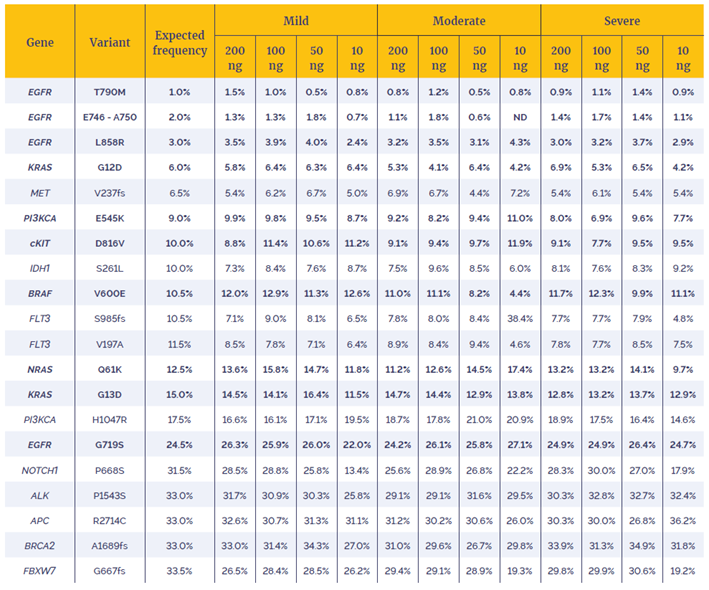 Table 2: VAFs of variants identified. Variants in bold have been confirmed by Horizon Discovery using droplet digital PCR, the presence of the remaining variants have been confirmed in the parental cell line. ND: not detected.
Table 2: VAFs of variants identified. Variants in bold have been confirmed by Horizon Discovery using droplet digital PCR, the presence of the remaining variants have been confirmed in the parental cell line. ND: not detected.
The data in Table 2 show that 99.6% of the expected variants were detected, even in the most severely damaged sample. Furthermore, the measured VAFs are close to the expected coverage with 91.25% of the 240 variants lying within 5 percentage points of their expected values.
For a more detailed view of the coverage, targeted regions were visualised with the Integrative Genomics Viewer (IGV) software. Using 200 ng of starting material, coverage of one SNV and two deletions are given for each of the three samples (Figure 6); Figure 7 shows the effect of decreasing the amount of input DNA.

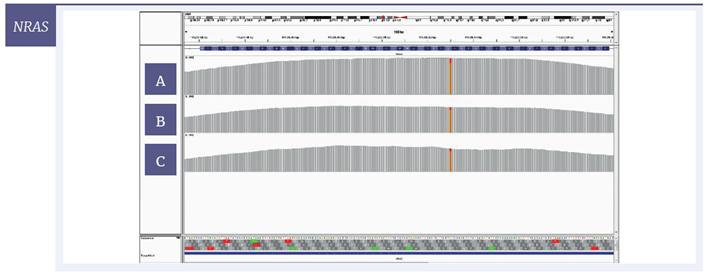
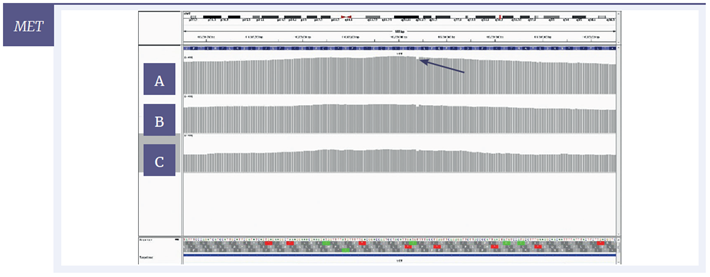
Figure 6: IGV plots of regions of the BRCA2, NRAS and MET genes with mild [A] , moderate [B] and severe [C] damage, 200 ng input DNA. All variants were detected at all levels of DNA damage. Deletions (BRCA2, MET) appear as single-nucleotide drops in coverage; the SNV (NRAS) appears as one line consisting of two colours with each colour representing a different nucleotide.
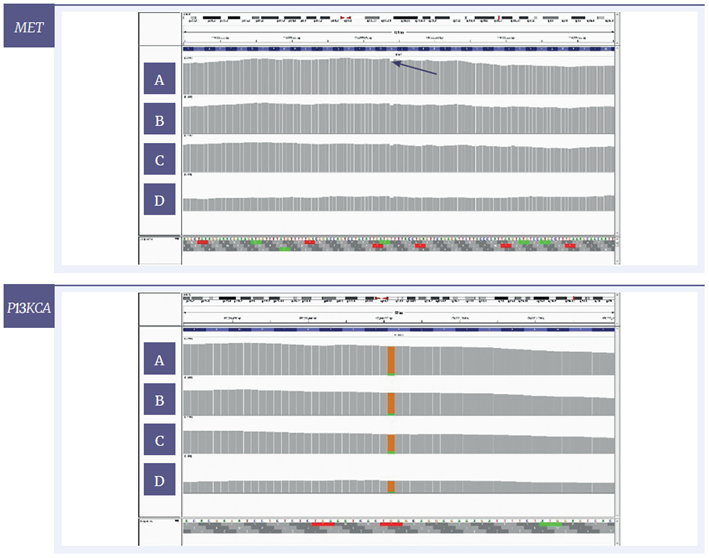 Figure 7: IGV plots of regions of the MET and PI3KCA genes of the sample with severe damage. The amounts of input DNA are 200 ng [A], 100 ng [B], 50 ng [C] and 10 ng [D]. Variants with a VAF of 6.5% and 9.0% respectively can be detected at all tested concentrations. MET - Y-axis set to 2500x, 2000x, 1000x and 500x for 200, 100, 50 and 10 ng respectively. PICK3CA- 2000x for 200 and 100ng and 750x for 50 and 10ng respectively.
Figure 7: IGV plots of regions of the MET and PI3KCA genes of the sample with severe damage. The amounts of input DNA are 200 ng [A], 100 ng [B], 50 ng [C] and 10 ng [D]. Variants with a VAF of 6.5% and 9.0% respectively can be detected at all tested concentrations. MET - Y-axis set to 2500x, 2000x, 1000x and 500x for 200, 100, 50 and 10 ng respectively. PICK3CA- 2000x for 200 and 100ng and 750x for 50 and 10ng respectively.
The plots in Figures 6 and 7 show that using the SureSeq FFPE DNA Repair Mix, reliably detect low-level variants in fcDNA even in as low as 10ng of severely damaged material. It also shows the high uniformity of coverage across the exon that can be achieved by using hybridisation-based enrichment.
In areas such as cancer research, the use of FFPE samples for sequencing has the potential to unlock a vast amount of information from tissue biopsies. The use of SureSeq FFPE DNA Repair Mix in addition to hybridisation-based enrichment has been shown to improve DNA yield and MTC, allowing greater confidence in calling low-frequency variants.
When studying 20 known variants, a 99.6% concordance was reported at all three levels of DNA damage (mild, moderate and severe). Of the measured allele frequencies, 91.25% of values were within 5 percentage points of the expected values. These results highlight that carrying out DNA repair in addition to hybridisation-based enrichment leads to better results in every step of the process – resulting in superior coverage, even when the quality or quantity of the starting material is low.
*The SureSeq FFPE DNA Repair Mix can only be purchased in conjunction with SureSeq NGS panels, not as a standalone product.
SureSeq™: For Research Use Only; Not for Diagnostic Procedures. This document and its contents are © Oxford Gene Technology IP Limited – 2021. All rights reserved. OGT™ and SureSeq™ are trademarks of Oxford Gene Technology IP Limited. The SureSeq NGS Library Preparation Kit was jointly developed between Oxford Gene Technology and Bioline Reagents Limited. MiSeq is a registered trademark of Illumina Inc.
 Visit USA site
Visit USA site Visit Canada site
Visit Canada site
