Inherited metabolic diseases comprise a large class of genetic diseases involving disorders of metabolism and while classed as rare disease, as a group are relatively common — as much as one in every 800 live births will have an inherited metabolic disorder1. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). The metabolic disorders are divided into a number of groups, each still containing many individual disorders, for example there are over 50 lysosomal storage disorders. Due to the large number of metabolic disorders there can be significant heterogeneity between them and understanding the complete genetic picture is made all the more important.
CytoSure® disease-specific arrays are designed to accurately identify small intragenic CNVs in genes associated with specific disorders. The content for the CytoSure Metabolic Disorder Research Array has been designed and optimised in collaboration with leading molecular genetics experts at Emory University.

Detect single or multiple exonic CNVs in the genes that matter

High-throughput, cost-effective analysis

Using industry-leading CytoSure Interpret® Software

Confident analysis and reporting
Inherited genetic disorders can be caused by a variety of chromosomal aberrations, including point mutations and small CNVs (Figure 2). Different methodologies are combined to accurately detect these changes, with one of the most successful combinations being targeted NGS and array comparative genomic hybridisation (aCGH)2. Using highly-targeted, exon-focused arrays as part of this combined approach has been shown to detect small CNVs (Figure 3).
The concept of using highly targeted gene-focused arrays to screen specific loci has been developed in collaboration with leading molecular genetics experts at Emory University and has now been implemented in multiple labs worldwide.
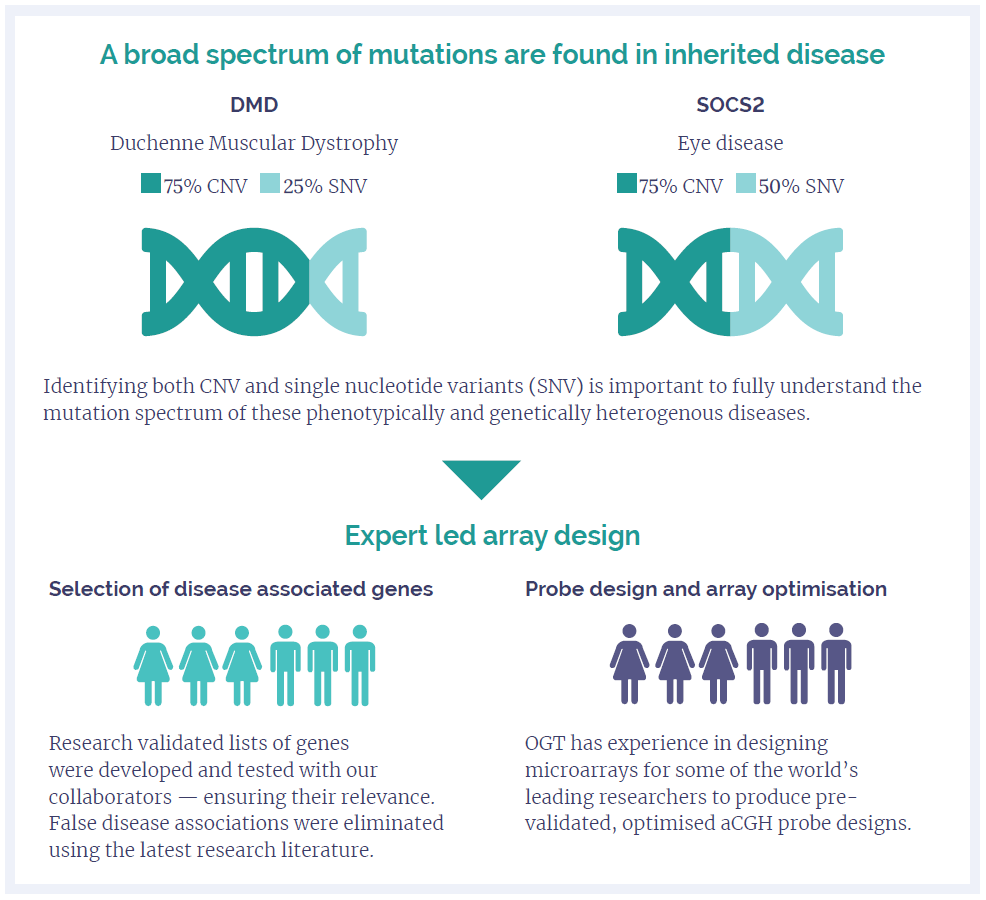
Figure 1: A broad spectrum of mutations are found in inherited disease
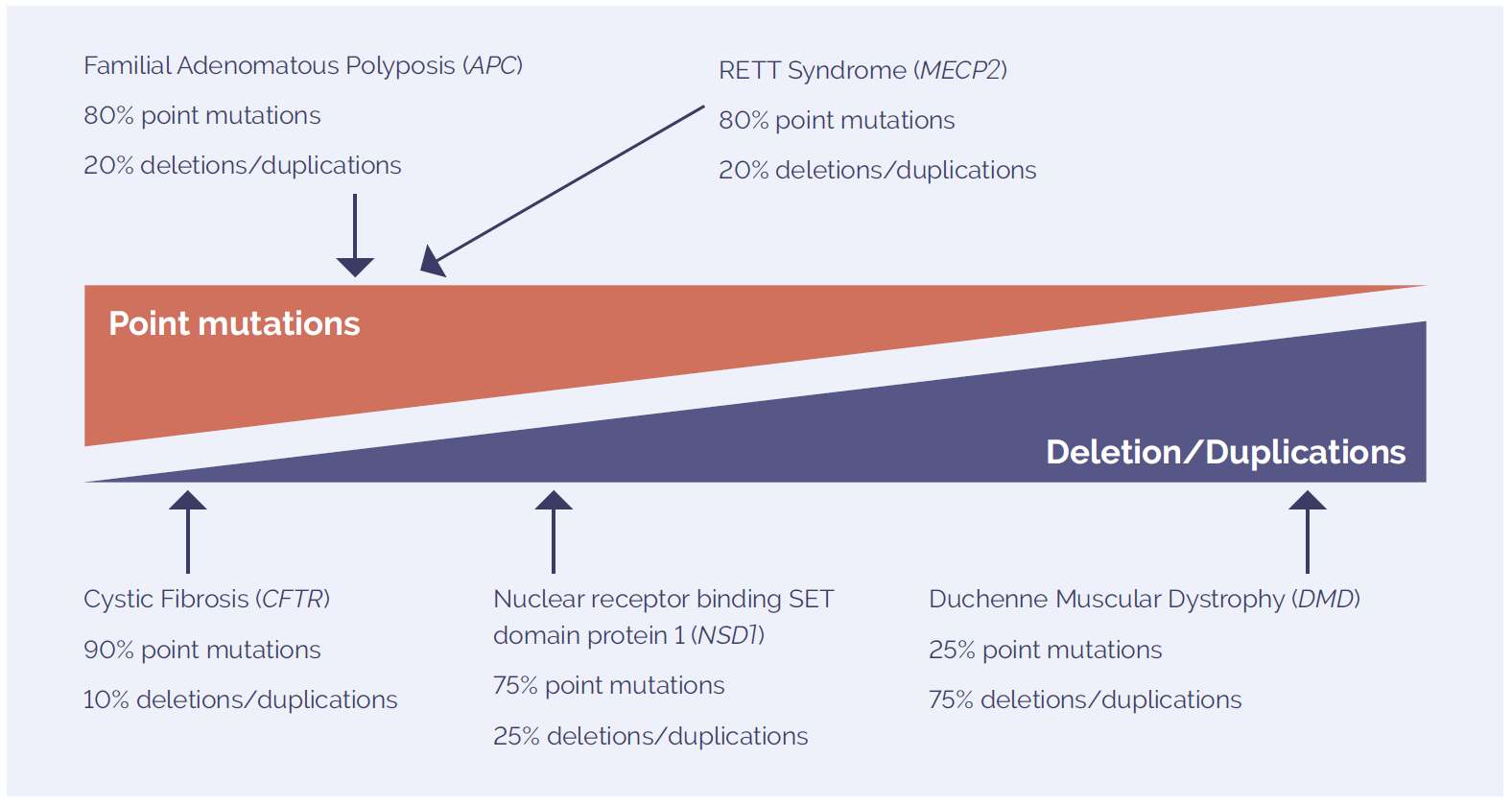
Figure 2: The mutation spectrum of molecular disorders includes point mutations and deletions and duplications. The prevalence of each type of variant is highly dependent on the disease studied.
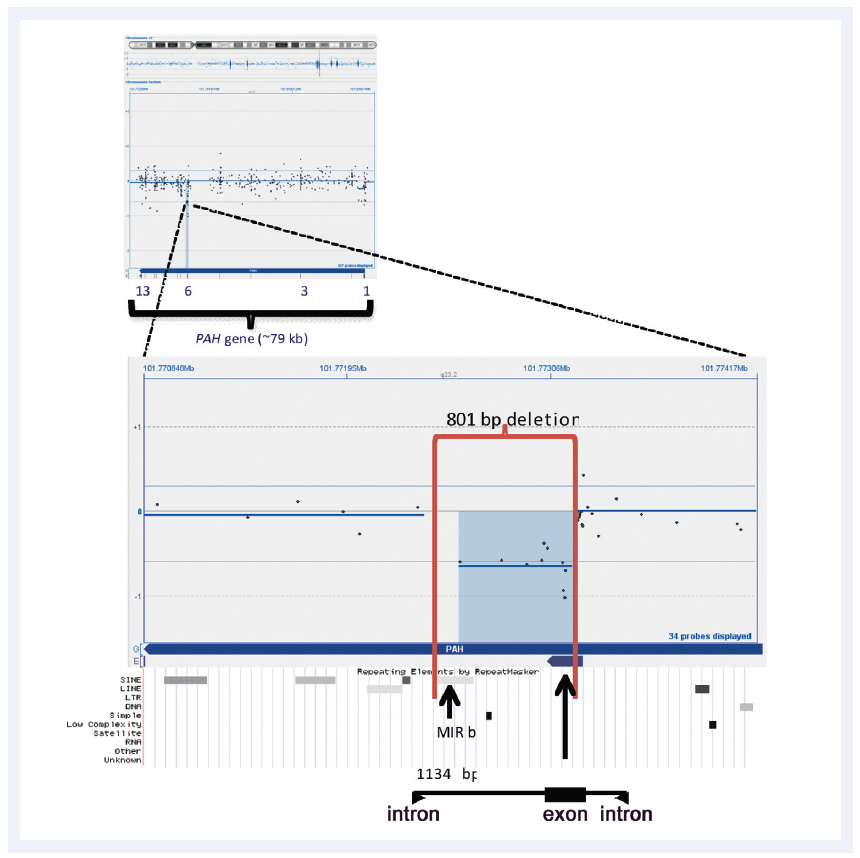
Figure 3: CytoSure Interpret Software enables easy detection and visualisation of small aberrations. Shown here is a 801bp micro-aberration causing partial deletion of exon 6 in the PAH gene (which in conjunction with a point mutation is causative of phenlyketonuria). Data kindly provided by Madhuri Hegde, Ph.D., FACMG, Emory University.
To detect these micro aberrations, highly optimised probes are required. The probes have been selected from OGT’s proprietary Oligome™ database — a database of over 25 million in silico optimised oligonucleotide probes. CytoSure arrays utilise 60-mer oligonucleotide probes, which have been shown to offer higher signal-to-noise ratios through increased specificity and sensitivity3. To further improve performance, during the design process multiple probes are designed for each of the target regions. These probes are tested in competitive hybridisation experiments and ranked based on technical performance — with only the best performing probes being chosen for the final array design. In addition, further experimental validation using clinical research samples has been performed by Emory University who collaborated in the design and optimisation of CytoSure Disease- Focused Research arrays.
Genetic analysis is typically done in batches to ensure cost-effective processing. However, collecting sufficient samples for rare disorders can significantly delay time to reporting. Through combining informative probes for multiple molecular disorders on a single array, CytoSure Disease-Focused Research arrays allow immediate and accurate processing of diverse samples, thereby reducing both time-to-results and cost.
Each CytoSure Disease-Focused Research array can be modified to create bespoke custom arrays, to suit your specific requirements. The customisation process is made easy with a dedicated project manager from our team of experienced computational biologists assigned to each new project. Additional content, specific to your research interest, can easily be selected from any existing array, our Oligome™ database of pre-optimised probes, or designed de novo to your specifications using our proprietary probe design pipeline. Delivery of your custom array only occurs once you are completely satisfied with the design. A number of array formats are available to suit your design and throughput requirements, with the most typical being 8x60k or 4x180k.
All CytoSure arrays are provided with CytoSure Interpret Software which is a powerful, easy-to-use package for the analysis of aCGH data. Innovative features such as the Accelerate Workflow enable standardised and automated data analysis, including automatic aberration detection and classification. In addition, extensive annotation tracks covering syndromes, genes, exons, CNV and recombination hotspots — each of which link to publicly available databases — can be used to provide results in context (Figure 4).
Figure 4: Fully customisable tracks in CytoSure Interpret Software simplify interpretation of aberrations.
All CytoSure arrays have been validated using CytoSure Genomic DNA Labelling Kits; these labelling kits have been developed and optimised to enable rapid delivery of high-quality results with excellent signal-to-noise ratios. The kits offer faster and simpler DNA labelling and clean-up than alternative enzymatic labelling procedures with improved data quality. Two kit formats are available; the CytoSure Genomic DNA Labelling Kit for 24 samples processed in tubes or the CytoSure HT Genomic DNA Labelling Kit for plate-based processing of up to 96 samples. For best results, CytoSure Genomic DNA Labelling Kits should be used with all CytoSure arrays to give the best derivative log ratio spread (DLRS) values and signal-to-noise ratios ensuring accurate detection of even the smallest aberration.
Increasing numbers of aCGH samples combined with higher-throughput array formats means that it is imperative to track samples throughout the labelling, hybridisation and analysis process to maintain sample identity. CytoSure Sample Tracking Spike-ins are uniquely designed to enable reliable sample tracking and easy identification of sample mix-up using OGT’s CytoSure Disease-Focused Research arrays and class-leading CytoSure Interpret Software.


 Visit USA site
Visit USA site Visit Canada site
Visit Canada site