

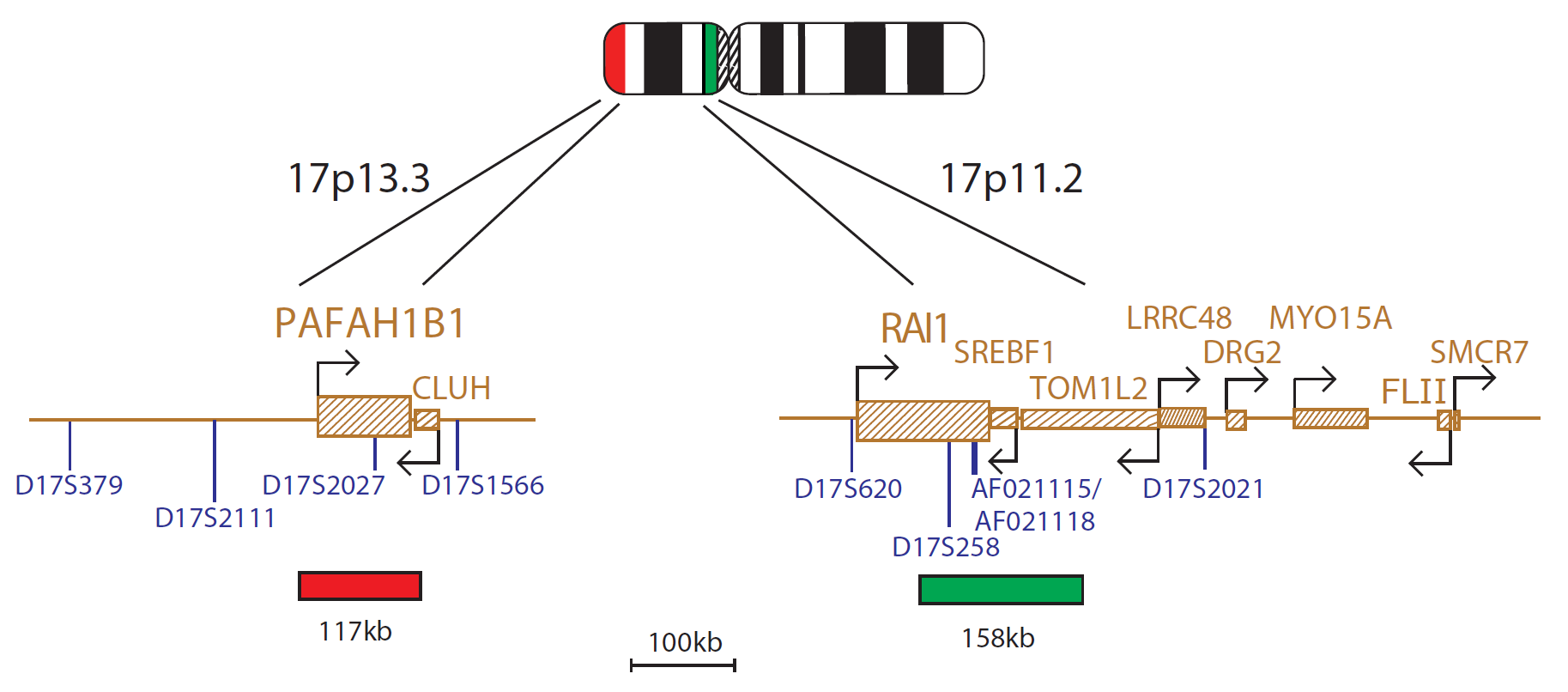
The Miller-Dieker (LIS1) probe is 117kb, labelled in red and covers the entire LIS1 (PAFAH1B1) gene. The Smith-Magenis (RAI1) probe is 158kb, labelled in green and covers the centromeric end of the RAI1 gene and includes the D17S258 marker. The two unique sequences act as control probes for each other and allow identification of chromosome 17.
Smith-Magenis syndrome (SMS) is a multiple congenital anomaly syndrome characterised by mental retardation, neurobehavorial abnormalities, sleep disturbances, short stature, minor craniofacial and skeletal anomalies, congenital heart defects and renal anomalies1,2.
It is one of the most frequently observed human microdeletion syndromes and is associated with an interstitial deletion of the chromosome band 17p11.22.
Molecular studies in SMS patients suggest a minimally deleted region (MDR) spanning approximately 700kb3,5, though the common deletion is around 4Mb in size4. The proximal boundary of the MDR is within a region of overlap between the FLII and LLGL1 genes, and the distal boundary within the PEMT gene3. Deletions or mutations in RAI1 (Retinoic Acid Induced 1) gene, which lies within the MDR, are associated with the syndrome3,5,6,7. RAI1 was shown to be the primary gene responsible for most features of SMS8,9.
Whilst deletion of the 17p11.2 region results in SMS, duplication of the same region results in a similar, yet distinct, disorder known as Potocki-Lupski syndrome10. Phenotypically this shares many similarities to SMS, though it is generally milder, but does have some unique clinical findings10.
The common duplication involves the same 4Mb region as the SMS deletion as both syndromes are mediated by non-allelic homologous recombination between flanking low copy repeat regions4.
Miller-Dieker syndrome (MDS) is a multiple malformation characterized by classical lissencephaly, a characteristic facial appearance andsometimes other birth defects11. It is associated with visible or submicroscopic rearrangements within chromosome band 17p13.3 in almost all cases12. Isolated lissencephaly sequence (ILS) consists of classical lissencephaly with no other major anomalies13. Submicroscopic deletions of chromosome 17p13.3 have been detected in almost 40% of these patients12.
MDS is considered a contiguous gene deletion syndrome where deletion of physically contiguous genes leads to the complex phenotypic abnormalities observed. The PAFAH1B1 (LIS1) gene is located at 17p13.3 and is recognised as the causative gene for the lissencephaly phenotype in both MDS and ILS14,15. Deletions in MDS patients always include the PAFAH1B1 gene, together with other telomeric loci to a distance in excess of 250kb14.

Microscope image
In vitro diagnostic (IVD)
→ English/Français/Italiano/Deutsch/Español
→ Polski
Research use only (RUO)
Find certificate of analysis documentation for our CytoCell FISH probes
Our lab has been using a wide range of CytoCell FISH probes for a number of years, and have been increasing this range all the time. The probes have clear bright signals and show good reproducibility. CytoCell provides fast delivery of catalogue probes, and are very responsive when we have any queries or problems with their products.

Bridget Manasse
Addenbrookes Hospital, Cambridge University Hosiptals NHS Foundation Trust, UK
In our hands, CytoCell FISH probes have proven to be of the highest quality with bright, easy to interpret signals, thus providing confidence in our results. OGT's customer support is outstanding, as their staff are extremely knowledgeable and truly care about their customers and their customers’ needs.

Jennie Thurston
Director of Cytogenetics, Carolinas Pathology Group, USA
I first came across CytoCell FISH probes in a previous lab I worked in and I was struck by the quality of the products. Since this time, I have been recommending and introducing CytoCell probes across all application areas — now they are the primary FISH probes used in our lab. They have an excellent range of products and their ready-to-use reagent format saves considerable time.

Elizabeth Benner
Medical Technologist, University of Arizona Health Network, USA
We have been working with CytoCell fish probes for two decades because of their excellent clarity and intensity regardless of the size of the probe. It is so clear and simple to detect.
Dr. Marina Djurisic
Head of Laboratory of Medical Genetics, Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic”, Serbia
The quality and consistency of CytoCell’s probes means I can trust the results, and my clients get their results in a timely manner.

Dr. Theresa C. Brown
Director, Cytogenetics Laboratory, Hayward Genetics Center, Tulane University School of Medicine, USA
It was very important for us to have more consistent results with our probes — easy-to-read bright signals and a range of vial sizes, which is much more cost-effective.

Janet Cowan, PhD
Director of the Cytogenetics Laboratory, Tufts Medical Center, USA
Not only do CytoCell offer an extensive range of high-quality FISH probes, the customer support is also excellent — providing fast access to all the probes I need. The probes are highly consistent with bright signals allowing easy scoring of results.
Dr. Eric Crawford
Senior Director, Genetics Associates Inc., USA
The quality and reproducibility of results using the CytoCell kit has been vital in accurately detecting co-deletions in our glioma investigations. We now have a cost-effective test that we can rely on that is also easy to use and interpret. We've been consistently impressed with this kit - not to mention the support offered by OGT's customer service, and have completely transitioned over to CytoCell probes.
Gavin Cuthbert, FRCPath
Head of Cancer Cytogenetics, Northern Genetics Servce, Newcastle, UK
 Visit USA site
Visit USA site Visit Canada site
Visit Canada site