

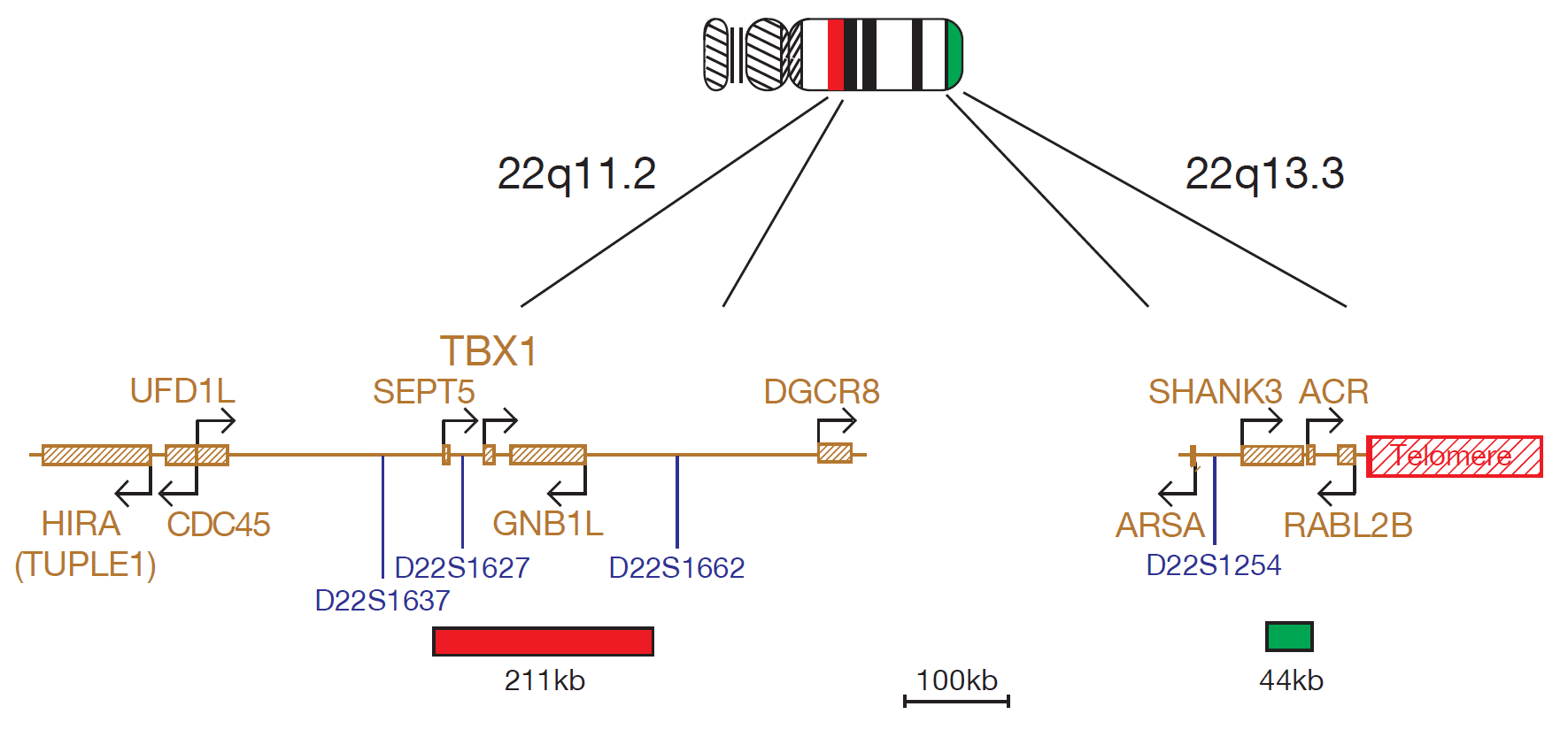
The TBX1 probe is 211kb, labelled in red, covers the entire TBX1 gene and includes the D22S1627 marker. The N85A3 (44kb), labelled in green, is located within 22q13.33 and covers the telomeric end of the SHANK3 gene, allowing for identification of the most distal 22q13.3 deletions. The two unique sequences act as control probes for each other and allow identification of chromosome 22.
DiGeorge Syndrome
DiGeorge syndrome1, and a variety of congenital malformation syndromes including Velocardiofacial syndrome (VCFS)2, share the deletion of chromosome 22 at 22q11.22,3,4,5.These chromosome 22 deletions are collectively coined CATCH22, a mnemonic that covers the clinical findings of Cardiac abnormality, Abnormal facies, Thymic aplasia, Cleft palate and Hypocalaemia/Hyperthyroidism due to a chromosome 22 deletion.
In addition, around 29% of nonsyndromic patients with isolated conotruncal defects have been shown to have a 22q11.2 microdeletion6. The incidence of these anomalies is estimated to be 1:4000 to 1:9700 live births7 and the deletion of 22q11.2 therefore represents one of the most common genetic defects.
A region of approximately 2Mb, referred to as the DiGeorge Critical Region (DGCR), is the most commonly deleted region and occurs in up to 90% of patients5,8,9. Within the DGCR, a minimal critical region of 300-480kb has been described10,11, containing several genes, including TUPLE1 (HIRA), TBX1, SLC25A1 (CTP) and CLTD.
22q13.3 Deletion Syndrome
The 22q13.3 deletion syndrome presents a recognisable phenotype characterised by hypotonia, delay or absence of expressive speech, global developmental delay, normal to accelerated growth and mild dysmorphic features12,13.
Some deletions of the terminal region of chromosome 22q are cytogenetically visible. However, a few cases of cryptic deletions have been reported12,14, suggesting that the actual incidence of 22q telomere deletion may be higher than previously thought.
Several observations of patients with 22q13.3 deletion showed that the SHANK3 (ProSAP2)20 gene, encoding a structural protein of the postsynaptic density of excitation synapses and expressed in the cortex and cerebellum of the brain15, was disrupted15,16,17 or deleted18, making it a candidate causative gene for this syndrome. The deletion varies dramatically in size from 130kb to 9Mb18,19,20. The use of 22q subtelomeric probes, distal to the ARSA gene, have therefore been recommended for examining all 22q13.3 deletions20,21.
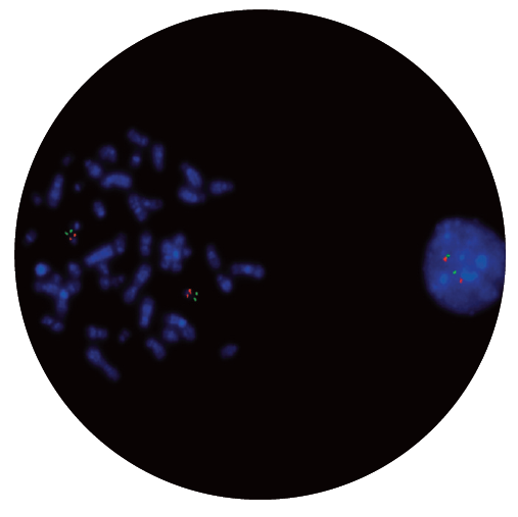
Microscope image
In vitro diagnostic (IVD)
→ English/Français/Italiano/Deutsch/Español
→ Polski
Research use only (RUO)
Find certificate of analysis documentation for our CytoCell FISH probes
Our lab has been using a wide range of CytoCell FISH probes for a number of years, and have been increasing this range all the time. The probes have clear bright signals and show good reproducibility. CytoCell provides fast delivery of catalogue probes, and are very responsive when we have any queries or problems with their products.

Bridget Manasse
Addenbrookes Hospital, Cambridge University Hosiptals NHS Foundation Trust, UK
In our hands, CytoCell FISH probes have proven to be of the highest quality with bright, easy to interpret signals, thus providing confidence in our results. OGT's customer support is outstanding, as their staff are extremely knowledgeable and truly care about their customers and their customers’ needs.

Jennie Thurston
Director of Cytogenetics, Carolinas Pathology Group, USA
I first came across CytoCell FISH probes in a previous lab I worked in and I was struck by the quality of the products. Since this time, I have been recommending and introducing CytoCell probes across all application areas — now they are the primary FISH probes used in our lab. They have an excellent range of products and their ready-to-use reagent format saves considerable time.

Elizabeth Benner
Medical Technologist, University of Arizona Health Network, USA
We have been working with CytoCell fish probes for two decades because of their excellent clarity and intensity regardless of the size of the probe. It is so clear and simple to detect.
Dr. Marina Djurisic
Head of Laboratory of Medical Genetics, Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic”, Serbia
The quality and consistency of CytoCell’s probes means I can trust the results, and my clients get their results in a timely manner.

Dr. Theresa C. Brown
Director, Cytogenetics Laboratory, Hayward Genetics Center, Tulane University School of Medicine, USA
It was very important for us to have more consistent results with our probes — easy-to-read bright signals and a range of vial sizes, which is much more cost-effective.

Janet Cowan, PhD
Director of the Cytogenetics Laboratory, Tufts Medical Center, USA
Not only do CytoCell offer an extensive range of high-quality FISH probes, the customer support is also excellent — providing fast access to all the probes I need. The probes are highly consistent with bright signals allowing easy scoring of results.
Dr. Eric Crawford
Senior Director, Genetics Associates Inc., USA
The quality and reproducibility of results using the CytoCell kit has been vital in accurately detecting co-deletions in our glioma investigations. We now have a cost-effective test that we can rely on that is also easy to use and interpret. We've been consistently impressed with this kit - not to mention the support offered by OGT's customer service, and have completely transitioned over to CytoCell probes.
Gavin Cuthbert, FRCPath
Head of Cancer Cytogenetics, Northern Genetics Servce, Newcastle, UK
 Visit USA site
Visit USA site Visit Canada site
Visit Canada site