
Want to download this as a PDF? Download now
Lyudmila Georgieva, Ezam Uddin, Jacqueline Chan, William Wright and Graham Speight
It has been reported that mutations in the CCAAT/enhancer binding protein alpha (CEBPA) and fms-related tyrosine kinase 3 (FLT3) genes are among the most common molecular alterations in acute myeloid leukaemia (AML). Two mutations on separate alleles of CEBPA with a specific combination of an N-terminal frameshift mutation on one allele and a C-terminal in frame mutation on the other allele are important for prognosis1,2. The prevalence of an internal tandem duplication (ITD) of the juxtamembrane domain-coding sequence and a missense mutation of D835 within the kinase domain of the FLT3 gene occurs in 15-35% and 5-10% of adults with AML, respectively3 . A quarter of patients with essential thrombocythemia or primary myelofibrosis carry a driver mutation of calreticulin (CALR). A 52-bp deletion (type 1) and a 5-bp insertion (type 2 mutation) are the most frequent variants4.
Further research to better characterize the involvement of these genes in acute myeloid leukaemia (AML) would be beneficial. NGS is one technology being explored for further research into myeloid disorders such as myeloproliferative neoplasms (MPNs) and AML but has been hampered by the difficulty in sequencing certain genes. Amongst the difficult to sequence genes are CALR, CEBPA and FLT3. The development of robust assays for CEBPA mutation analysis is challenging due to the high GC content of the gene (75% in the coding region), the presence of a trinucleotide repeat region, the complexity of the mutations, and the frequent occurrence of mutations in mononucleotide repeats. Genes such as FLT3 are challenging to target because they contain complex repetitive elements that can be long and are generally masked in most panel designs. CALR sequencing is challenging due to the presence of low complexity regions making the detection of insertions and deletions difficult.
The SureSeq™ hybridization-based approach was used throughout this study; the workflow of this is outlined in Figure 1.
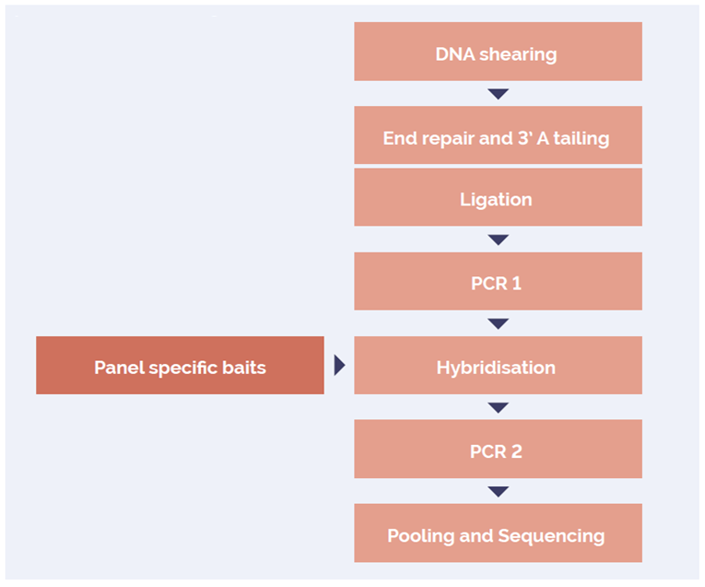 Figure 1: OGT SureSeq workflow. The SureSeq workflow allows users to go from extracted DNA to sequencer in 1.5 days with minimal handling time.
Figure 1: OGT SureSeq workflow. The SureSeq workflow allows users to go from extracted DNA to sequencer in 1.5 days with minimal handling time.
We have utilized a hybridization-based enrichment approach in combination with a SureSeq myPanel™ NGS Custom AML Panel.
The panel was used to confirm variants in research samples* containing variants in each of these difficult to sequence genes. Sequencing was conducted on a MiSeq® using a V2 300 bp cartridge (Illumina).
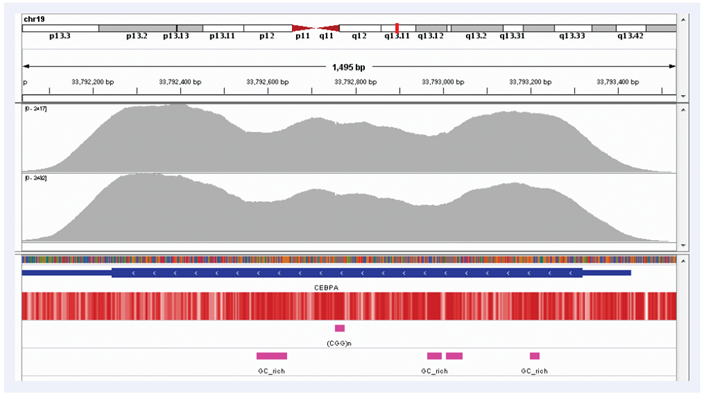 Figure 2: Excellent uniformity of coverage of the CEBPA gene (average depth~2000x). Depth of coverage per base (grey). GC percentage (red). Repeat regions and GC-rich regions (pink).
Figure 2: Excellent uniformity of coverage of the CEBPA gene (average depth~2000x). Depth of coverage per base (grey). GC percentage (red). Repeat regions and GC-rich regions (pink).
 Figure 3: Detection of FLT3-ITDs with different sizes. Wild-type sample (bottom panel).
Figure 3: Detection of FLT3-ITDs with different sizes. Wild-type sample (bottom panel).
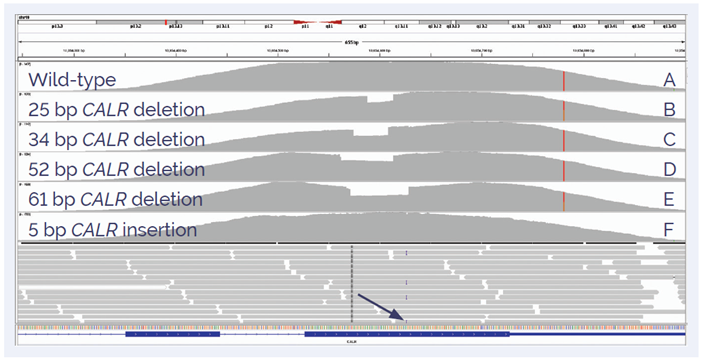 Figure 4: Detection of insertions and deletions in exon 9 of CALR. Wild-type sample (panel A) is compared to a 25 bp (panel B), 34 bp (panel C), 52 bp (panel D), 61 bp somatic deletions (panel E) and 5 bp insertion (panel F).
Figure 4: Detection of insertions and deletions in exon 9 of CALR. Wild-type sample (panel A) is compared to a 25 bp (panel B), 34 bp (panel C), 52 bp (panel D), 61 bp somatic deletions (panel E) and 5 bp insertion (panel F).
 Figure 5: Detection of insertions and deletions in CEBPA.
Figure 5: Detection of insertions and deletions in CEBPA.
*Research samples provided by the National Genetics Reference Laboratories - Wessex, University of Virginia, University of Newcastle and UK NEQAS.
SureSeq™: For Research Use Only; Not for Diagnostic Procedures.
 Visit International site
Visit International site Visit USA site
Visit USA site International
International
