
Laura Semenuk BSc MLT, Kassandra Sims BSc MLT, Alissa Palazzolo BSc MLT, Dr. Michael Rauh MSc, PhD, MD, FRCPC, Dr. Graeme Quest MD, MSc, FRCPC
Hematological malignancies cover a diverse and heterogeneous range of diseases, of which the two primary lineages are myeloid and lymphoid neoplasms, across a spectrum of leukemias, myeloproliferative disorders, myelodysplastic syndromes and lymphomas. The genomic characterization of these diseases typically requires the identification of a multitude of disease-driving variants, from single nucleotide variants to multi-nucleotide variants (such as insertions and deletions) and larger copy number variants.
At Kingston, we typically find that a laboratory diagnosis pathway for myeloid and lymphoid neoplasms encompasses several separate and/or sequential analyses to fully assess the full range of genomic variants. The limitations of this approach include the limited scope of singular targeted tests, the capital required for the implementation of multi-technology workflows. Additionally, considerations for laboratory personnel such as the strain of repeated techniques, can provide a further barrier. The benefits of a more separate/sequential analysis workflow can be access to rapid, precise and low-cost assays that can be linked to known therapeutics upon diagnosis.
A combined testing approach for both lineages, which uses comprehensive next-generation sequencing (NGS), could potentially offer the same benefits while providing a more in-depth genomic profile of each sample, such as stronger characterization of structural variants. This would ultimately provide a better clinical understanding of disease, for instance identifying novel disease-drivers or enabling sample stratification based upon the variants detected. We are conscious however that there are some barriers to this approach such as the need for stringent analysis of sequencing data and requirements for data storage and these would need to be accounted for to adopt this methodology.
We undertook the validation of a comprehensive NGS approach for the identification of multiple types of genomic drivers, using the SureSeq myPanel™ service to create a custom panel for the analysis of both myeloid and lymphoid neoplasms.
Peripheral blood (PB) and bone marrow (BM) specimens were obtained from a diverse range of suspected clinical subtypes as shown in Table 1. Upon sample acquisition, DNA was extracted using either a column-based approach [PB samples] (QIAamp DNA Blood Mini Kit, Qiagen, USA) or salt-extraction method [BM samples] (Gentra Puregene DNA Kit, Qiagen, USA). Library preparation was performed using the OGT Universal NGS Workflow Solution (described below).
To create a custom SureSeq myPanel, the targeted NGS SureSeq™ CLL + CNV Panel and SureSeq Pan- Myeloid Panel from OGT were combined and further customized to include additional clinically relevant hematological targets and backbone coverage for chromosomes of interest, such as BCL2, NOTCH2 and Trisomy 8. The final panel was able to detect CNVs (specifically large CNVs in chromosomal backbone regions and exon/whole gene level CNVs in targeted regions), SNVs, Indels, ITDs and PTDs for gene markers, where relevant.
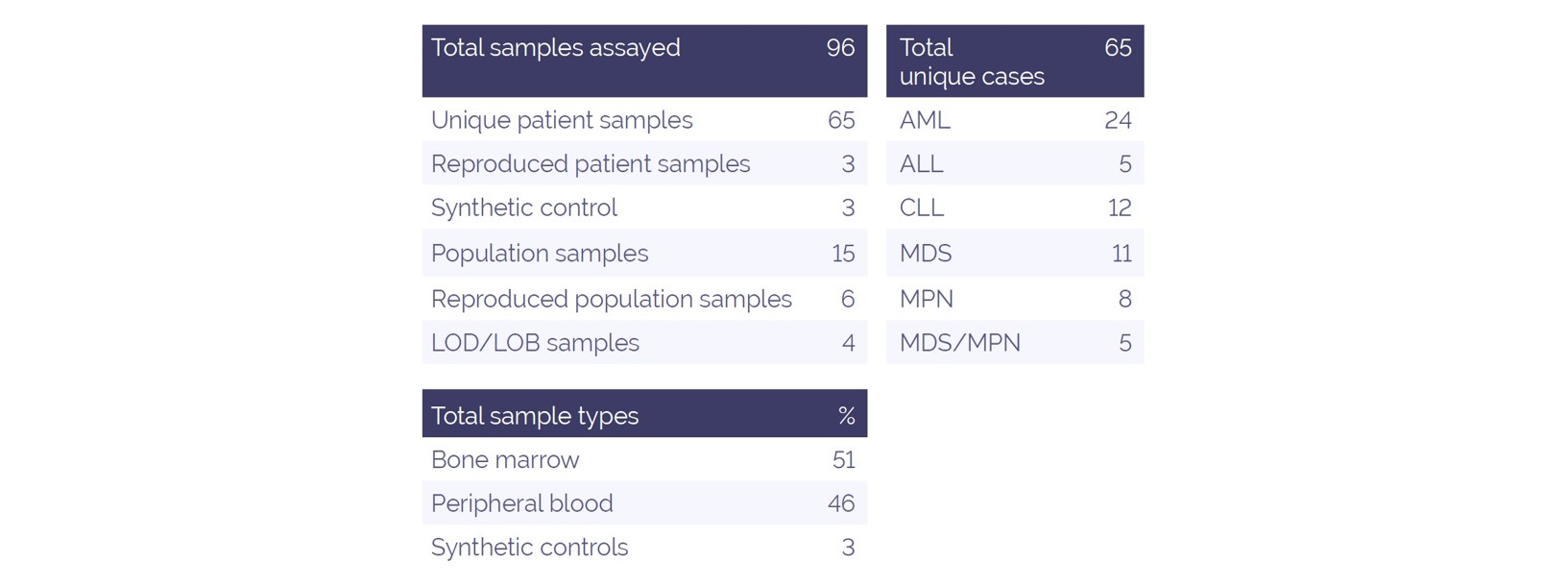
Table 1. Sample information
The OGT Universal NGS Workflow Solution was used throughout this study (Figure 1). The approach offers a streamlined NGS library preparation protocol with unique dual indexing (UDI), followed by hybridization-based target enrichment. This study utilized 500ng DNA per sample for library preparation in conjunction with the panel described above. Sequencing was performed in batches of 32 samples with a P1-300 cycle cartridge (Illumina, USA) using a NextSeq® 2000 (Illumina, USA).
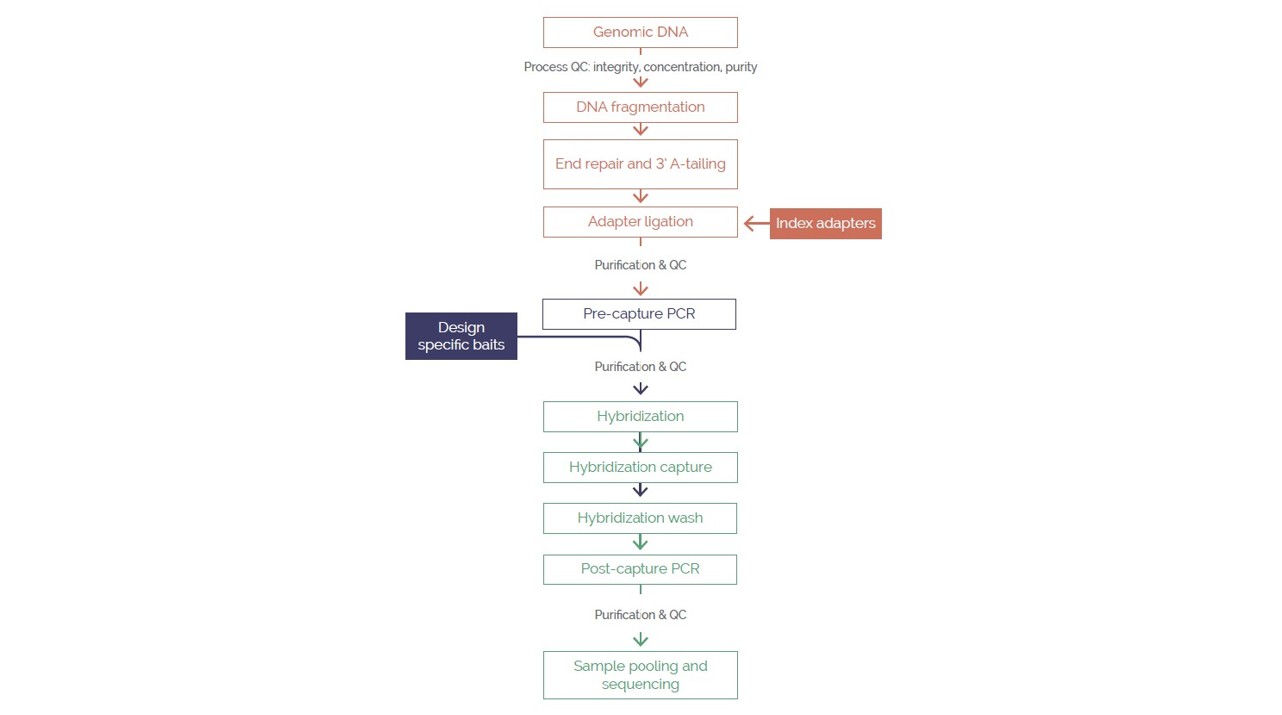
Figure 1. The OGT Universal NGS Workflow Solution
Sequencing data analysis was performed using OGT’s proprietary Interpret software, including read mapping, error correction, coverage calculation and variant calling, with alignment to GRCh37. CNV calling was further enhanced with an updated Interpret pipeline during validation of this panel.
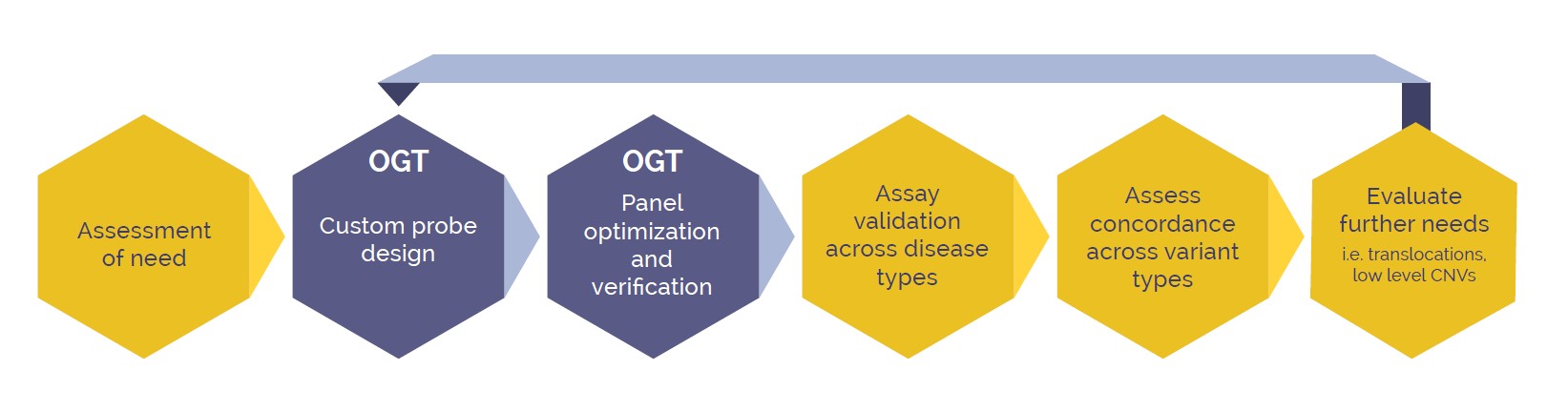
Figure 2. Development pathway of a new NGS assay, working in conjunction with specialists at OGT to create a customised targeted, NGS panel
To generate a custom NGS panel we worked with OGT to combine the SureSeq CLL + CNV Panel and the SureSeq Pan-Myeloid Panel. Additional gene content was built into the panel from a combination of OGT’s pre-existing content library and custom bait designs (Figure 2). OGT developed a fully pre-optimized panel capable of detecting SNVs, Indels, ITD, PTDs and CNVs (including exon level and whole gene CNVs) for which OGT verified technical performance. From there, the panel was placed in our hands to perform preliminary testing against expected performance for variants and disease types in our own laboratory workflow. Once approved the final panel content was generated and deployed for validation.
As part of the custom panel development, OGT conducted optimization of targeted content as per our requirements. STAG2 is a good example of a target that typically has low coverage compared to other targets due to AT-rich regions. Figure 3 highlights the improvement in coverage for 3 example exons attained by OGT through an optimized baiting strategy. Across all optimized STAG2 target regions an average 22.3% improvement in coverage was achieved.
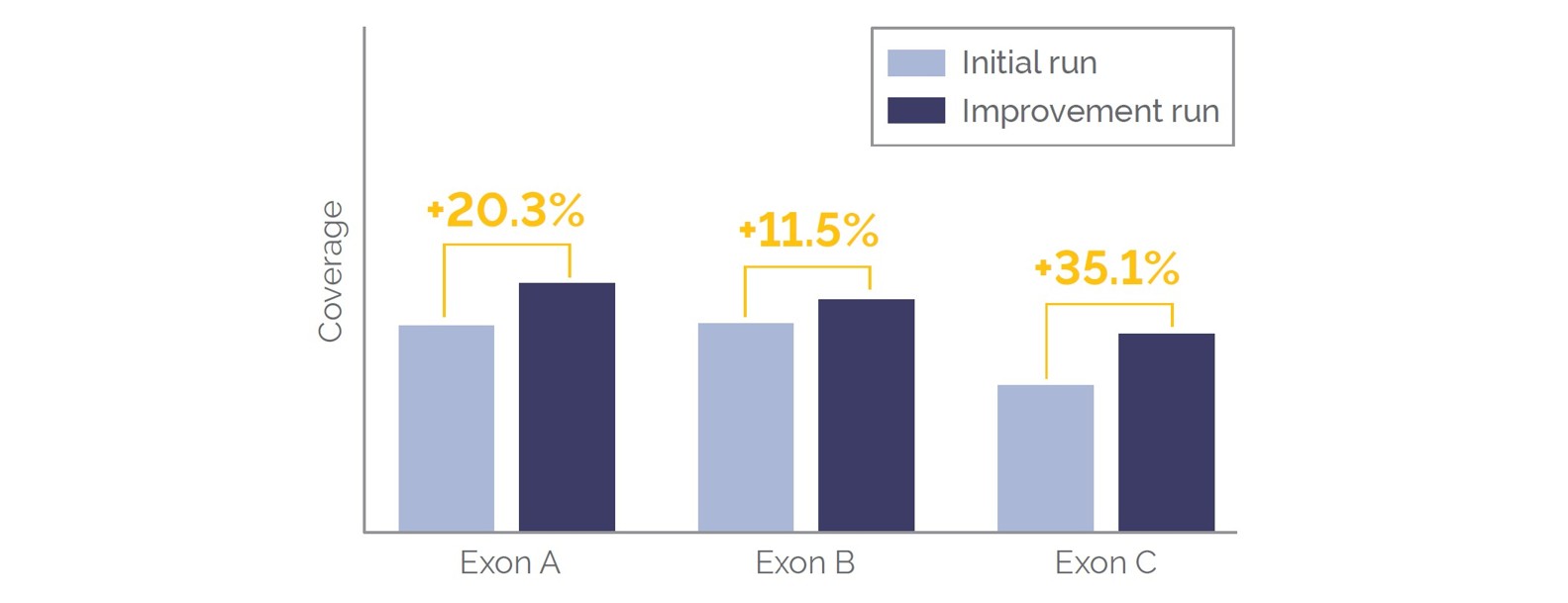
Figure 3. Coverage improvements in STAG2 exons following optimization of baiting strategy by OGT.
As part of the development pathway, we generated sequencing data for a validation cohort of 96 independently prepared libraries, comprised of 65 unique samples, with 3 and 6 repeated for intra-run and inter-run repeatability, respectively. Average total reads were 6.9 million, with a mean read coverage of 1040 per specimen. An average mapped read percentage of 99.9% was obtained. Library failure rate due to poor sequencing coverage or a shifted insert size was found to be 5%.
CNV detection with the first iteration of bioinformatic analysis demonstrated an analytical sensitivity and specificity at 84% and 94% respectively.
Initially, false negative results were found for mosaic level CNV findings with a copy number clonality less than 50%, as compared to karyotype findings. Subsequent optimization of the bioinformatic analysis, with a previously sequenced male negative control sample set, allowed for an increased resolution of copy number clonality findings down to 30%, as compared to karyotype findings.
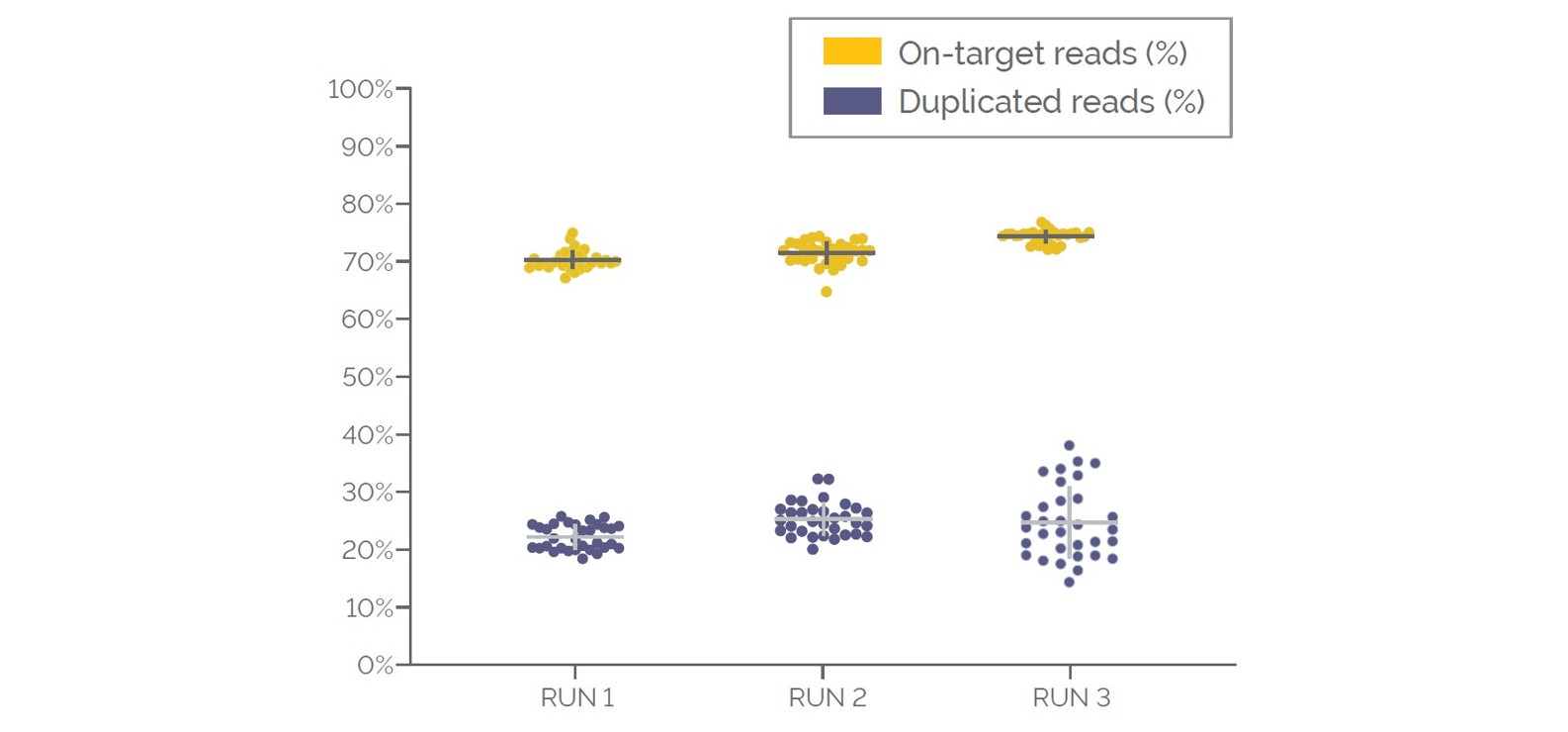
Figure 4. Coverage quality metrics for the validation cohort. Data displayed in the order the samples were processed, for the percentage of on-target reads and duplicated reads.
Results from the analysis of the individual 65 cases resulted in the detection of pathogenic variants in 78.5% of these cases (Figure 5). Variants included inframe, missense and truncating single and multi-nucleotide mutations. Additionally, 36 cases presented with multiple pathogenic variants. Analytical sensitivity and specificity for SNV/MNV detection was 98% and 100%, respectively.
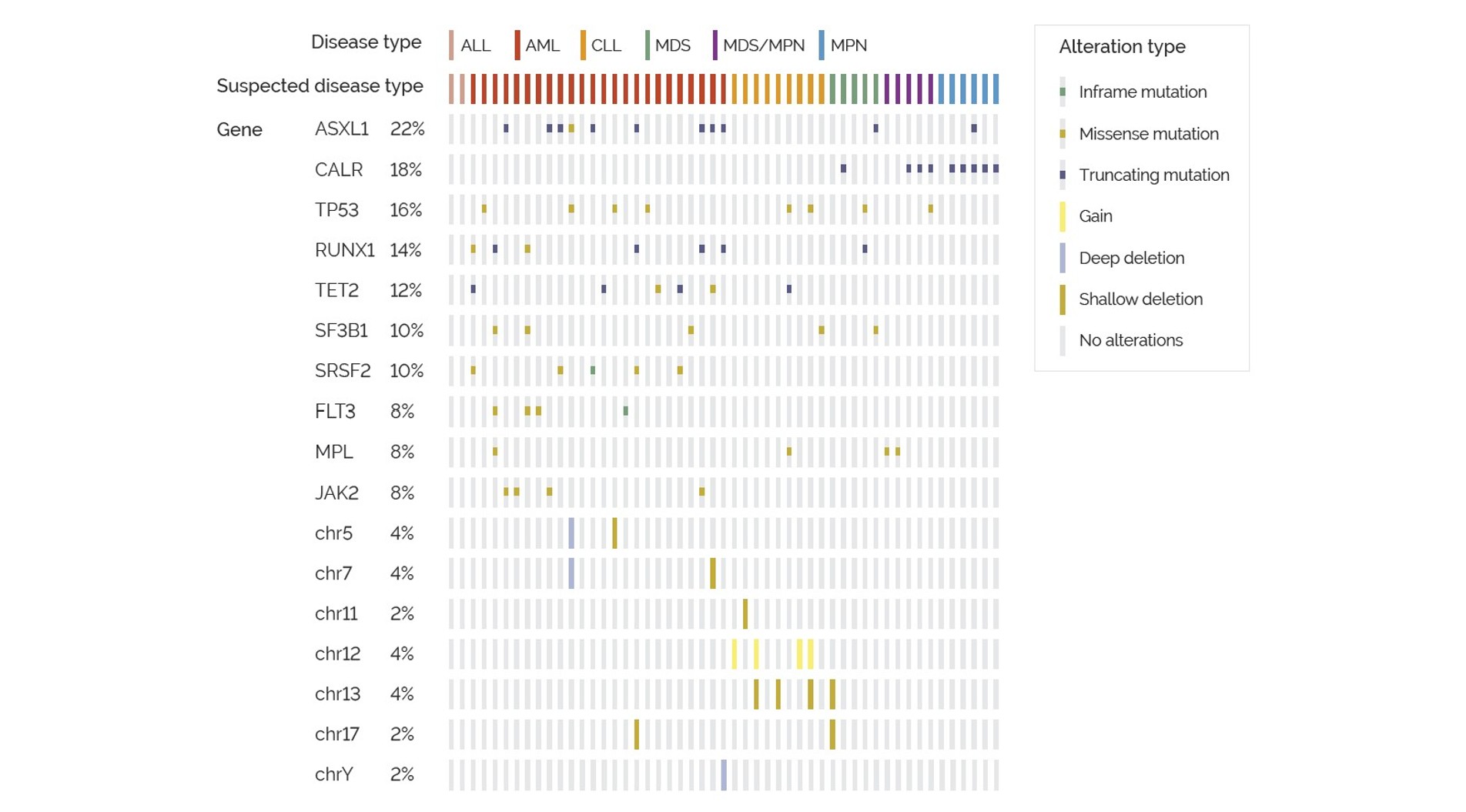
Figure 5. Oncoprint of the most frequent pathogenic variants within the validation cohort, by disease type and type of genetic alteration.
A targeted, next-generation sequencing panel has the technical capability to detect a multitude of genomic variants within a comprehensive assay, providing diagnostic, prognostic and therapeutic value, with efficiency improvements and increased fiscal responsibility.
Tailor your NGS panel to meet your specific analytical needs, ensuring maximum efficiency and focus on the most relevant insights for your research. Simply select your desired targets from OGT’s regularly updated, expert curated library of pre-optimized cancer content and we’ll work with you to deliver a technically verified panel that meets your exact requirements.
Figure 6. SureSeq myPanel workflow
Call +44 (0)1865 856800 Email contact@ogt.com
Send us a message and we will get back to you
 Visit International site
Visit International site Visit USA site
Visit USA site