
Jacqueline Chan, Juliette Forster, Aysel Heckel, Venu Pullabhatla, Dave Cook, Graham Speight
Formalin-fixed, paraffin-embedded (FFPE) storage is a standard method for archiving samples from solid tumors. It ensures the preservation of the ultrastructure of tissues and prevents degradation through formation of chemical links between macromolecules, for example between and within DNA molecules. FFPE samples contain a wealth of information which can be used to study cancer development and progression. Next generation sequencing (NGS) offers the capability of unlocking this information through the simultaneous study of multiple types of mutations in cancer-associated genes for a number of applications1. However, formalin treatment can significantly compromise the quality and amount of nucleic acids available for genomics research. As such it is technically challenging to examine the true genetic complexity present in a sample.
In this study DNA reference standards with different levels of formalin-induced damage were hybridized and sequenced with a SureSeq™ custom NGS panel in conjunction with the SureSeq FFPE DNA Repair Mix*. We assessed the impact of the repair mix on three levels of formalin compromised DNA (fcDNA) - ‘mild’, ‘moderate’ and ‘severe’, at 4 different DNA input amounts down to 10 ng. We then compared the uniformity and coverage of the enriched targets. We also assessed the concordance to the allele frequencies of the variants in the reference standards.
We tested three reference standards of fcDNA (provided by Horizon Discovery†) with ‘mild’, ‘moderate’ and ‘severe’ damage2. The samples were investigated in duplicate before and after repair with SureSeq FFPE DNA Repair mix; the amounts of input DNA were 200, 100, 50 and 10 ng (Figure 1). All samples were sheared using a Covaris S220 focused-ultrasonicator and prepared using the SureSeq NGS Library Preparation Kit (cat. no. 500070).
Enrichment by hybridization was completed with a SureSeq custom NGS 8.7 Kb custom hot-spot panel designed to target the variants present in the reference standards (see Table 1 for variants targeted). The subsequent post-capture libraries were sequenced on an Illumina MiSeq® using a v2 300 cycles kit (cat. no. MS-102-2002). 16 samples were run on a MiSeq lane.
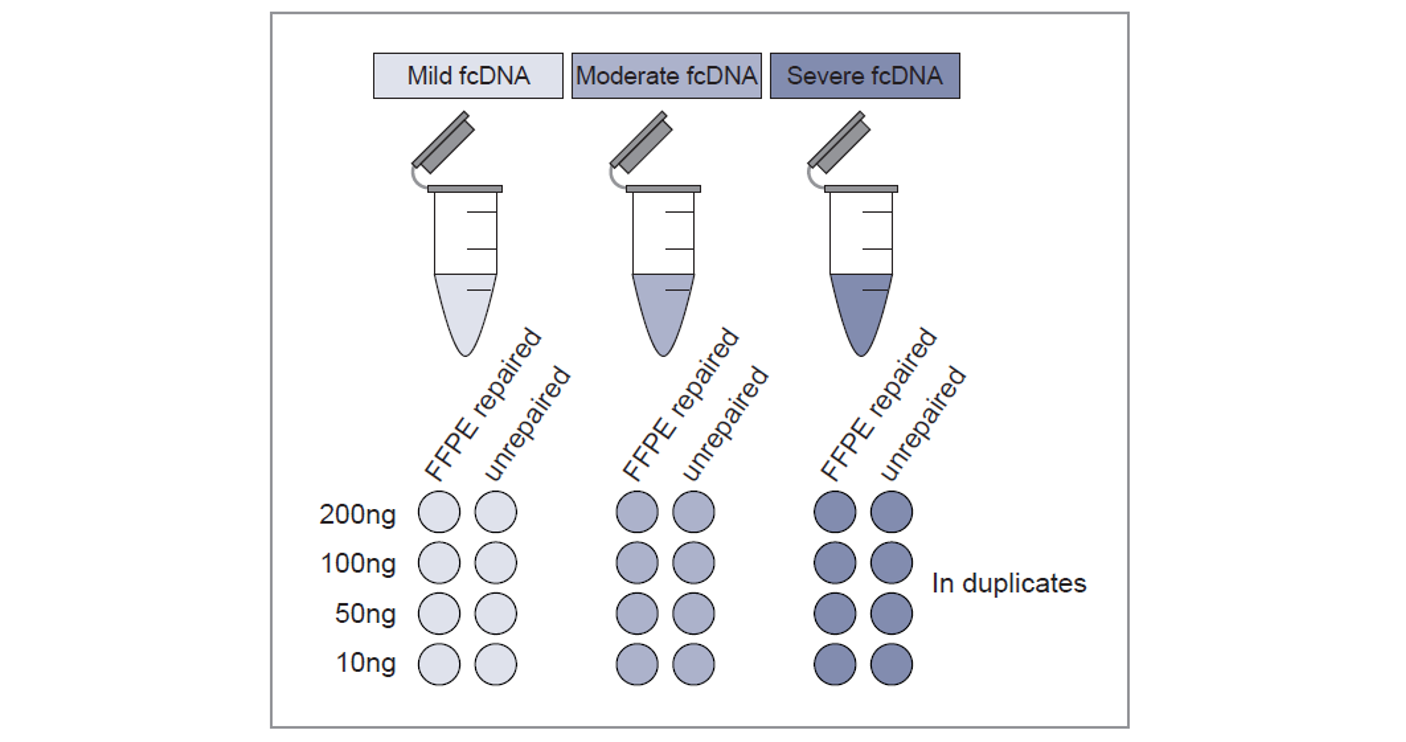
Figure 1: Experimental design. A total of 48 samples were sequenced to study the effect of DNA quality, input amount and DNA repair.
The SureSeq hybridization-based approached was used throughout this study; the workflow of this is outlined in Figure 2.
Use of target-capture allows the removal of PCR duplicates which can obscure the minor alleles present within a sample.
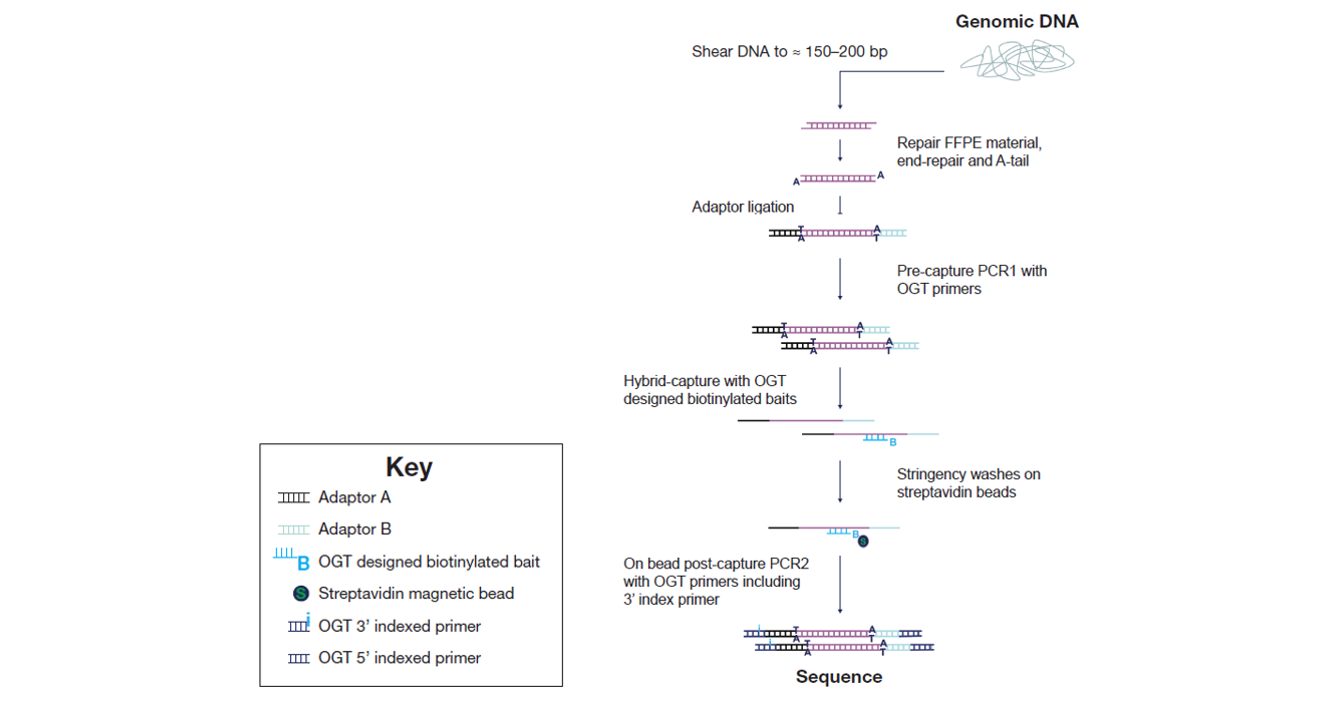
Figure 2: OGT SureSeq workflow. The SureSeq workflow allows users to go from extracted DNA to sequencer in 1.5 days with minimal handling time.
We found pre-treatment with the FFPE DNA Repair Mix improved library yields (Figure 3), which in turn led to an improvement in mean target coverage (Figure 4). Yield improvements varied from 1.2x in mildly compromised samples to 1.5x in severely damaged samples.
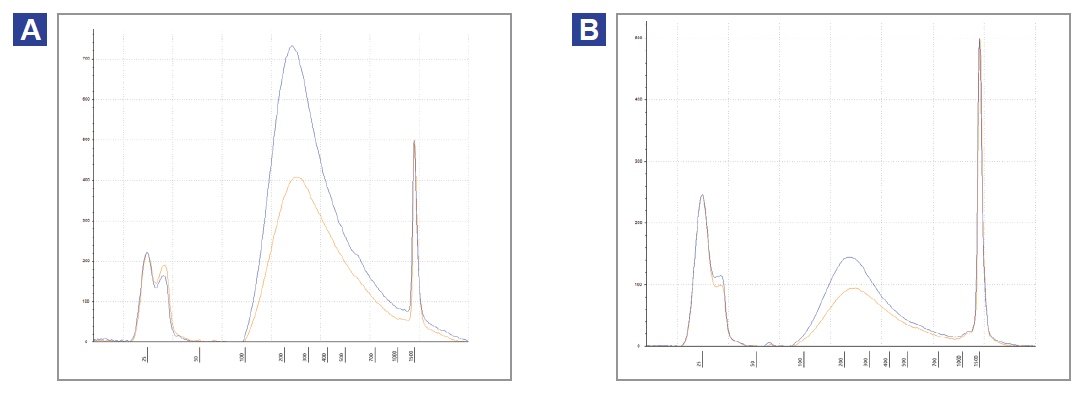
Figure 3: Agilent TapeStation traces of pre-capture libraries. Illumina-compatible libraries were prepared with severely damaged fcDNA that was treated with SureSeq FFPE DNA Repair Mix (blue), or was untreated (orange). The amount of input DNA were (A) 200 ng and (B) 50 ng.
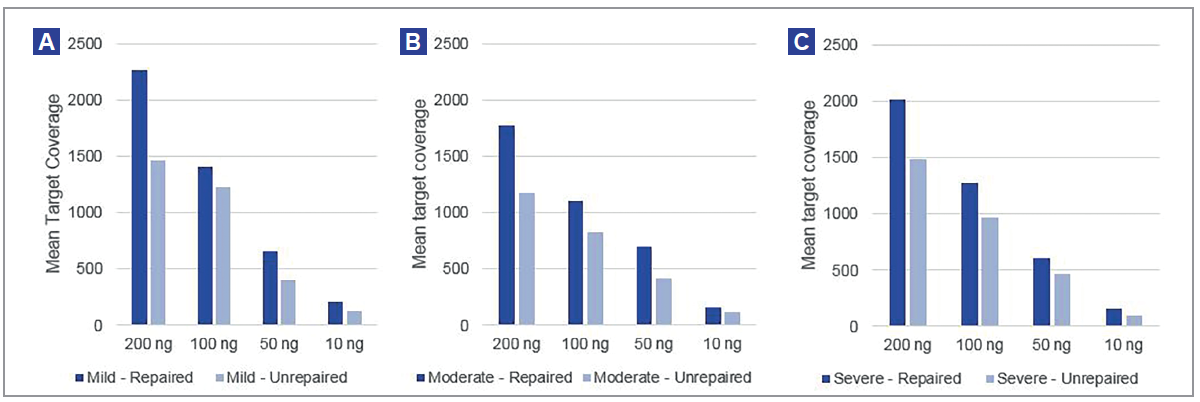
Figure 4: Improvement in mean target coverage can be observed across all amounts of starting material and all levels of DNA damage. (A) – mild fcDNA; (B) – moderate fcDNA; (C) – severe fcDNA.
The amount of archived sample tissue available can be limited and may only contain a low percentage of tumor cells of interest, therefore assays need to demonstrate strong sequencing performance at low input amounts. High depth and uniformity of coverage enables the accurate detection of low frequency mutations.
Samples treated with FFPE Repair Mix also demonstrated good sequencing metrics at low input amounts, maintaining a high and uniform level of coverage over a range of input amounts (Figure 5).
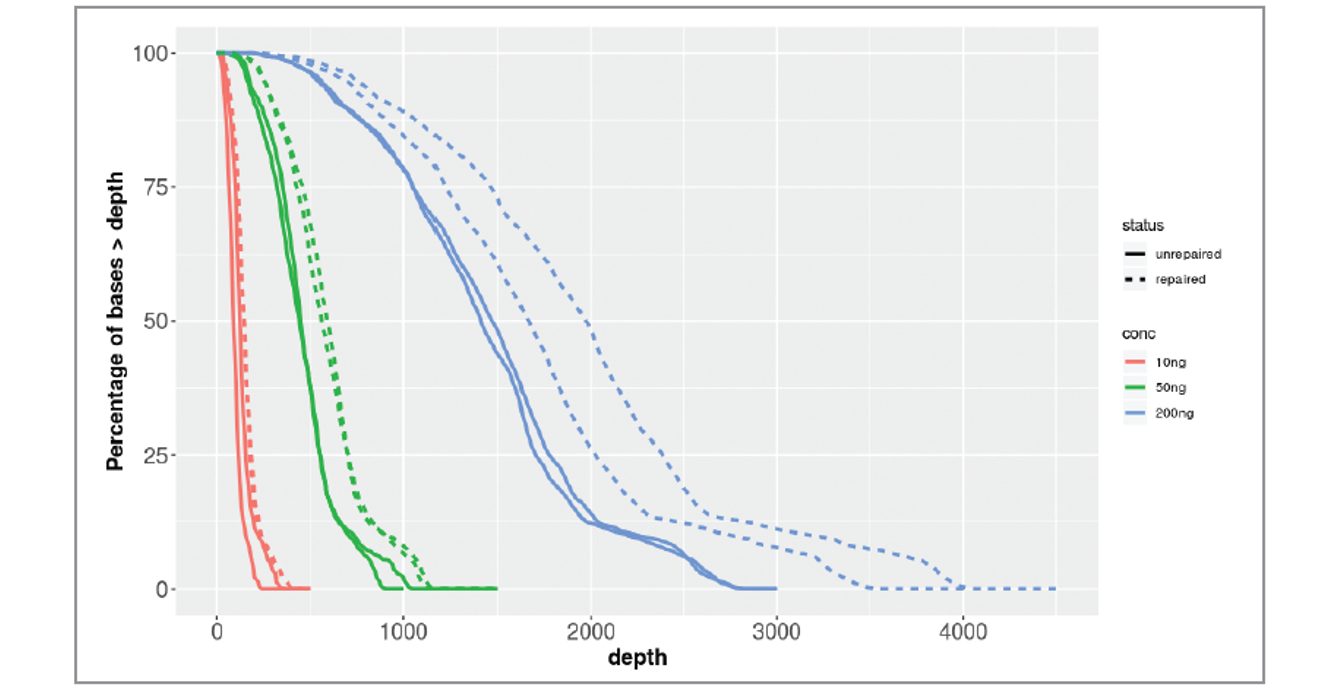
Figure 5: Plot of % bases at greater than x depth for treated (dotted lines) and untreated severely fcDNA (solid lines) samples using 10 (red), 50 (green), and 200 (blue) ng input.
The quantity and quality of sequencing data has a direct impact on the confident identification of variants, in particular, low frequency somatic variants. We found the number of supporting reads increased in repaired samples as shown in Figure 6 and reduced the number of false positive calls.
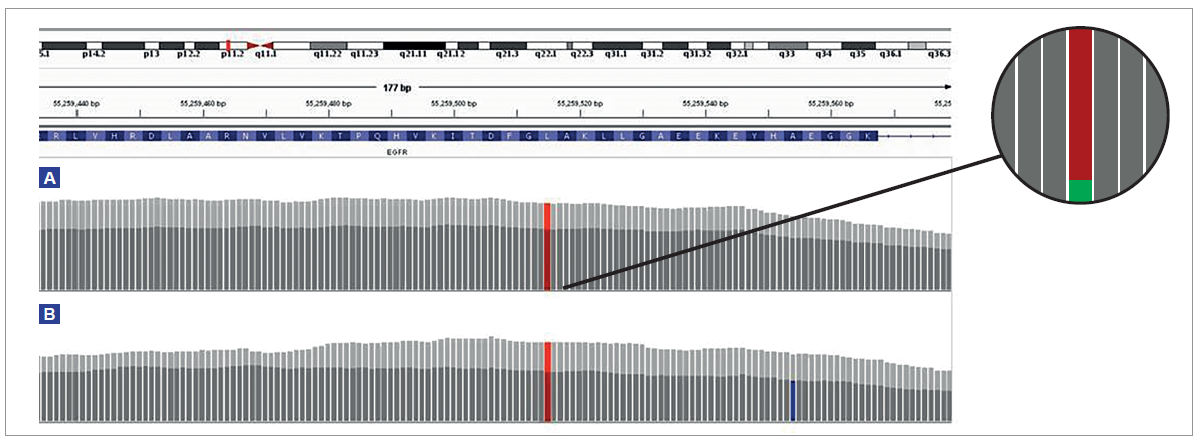
Figure 6: Comparison of the depth of coverage over a 3% EGFR L858R mutation in a severely formalin-compromised sample treated with FFPE repair mix (light grey) and not treated (dark grey). At 200 ng input (A), following treatment, the total depth increased by 43%, from 2707 to 3860, and the number of reads supporting the variant increased from 85 to 112, an increase of 32%. Panel shows expanded illustration of the reads supporting the reference (red) and reads supporting the variant (green). At 10 ng input (B), following treatment, the total depth increased by 58%, from 216 to 341, with the number of supporting reads increasing from 5 to 13 (160%). The combination of low input and poor quality DNA increases the likelihood of false positive mutations (blue).
The custom hot-spot panel was designed to capture 20 variants present in the reference standards: 15 single nucleotide variants (SNVs) and 5 deletions with variant allele frequencies varying between 1 and 33%. We found 100% concordance in repaired samples with >500x coverage, including three sub-5% EGFR variants. Overall 99.6% of the expected variants were detected with 91.25% of the 240 variants lying within 5 percentage points of the expected values (Table 1).
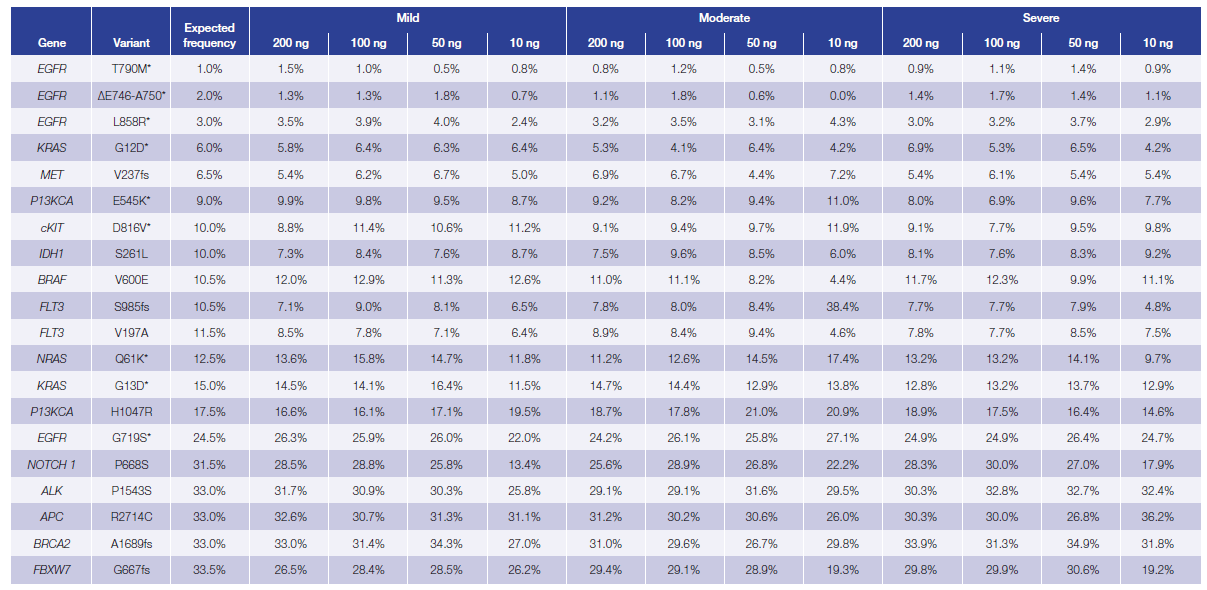
Table 1: Difference between the expected and observed allele frequency in Horizon Discovery’s fcDNA standards treated with FFPE Repair Mix (unrepaired data not shown). The variants were identified using OGT’s Interpret™ software. Mean of duplicates shown.
*Variants confirmed by Horizon Discovery droplet digital PCR, the presence of the remaining variants were confirmed in the parental cell line.
We found the correlation between expected and observed values was improved in samples treated with FFPE Repair Mix (Figure 7). The greatest improvement in accuracy of MAF value was found in severely formalin-compromised samples, in particular when starting with 10 ng of DNA.
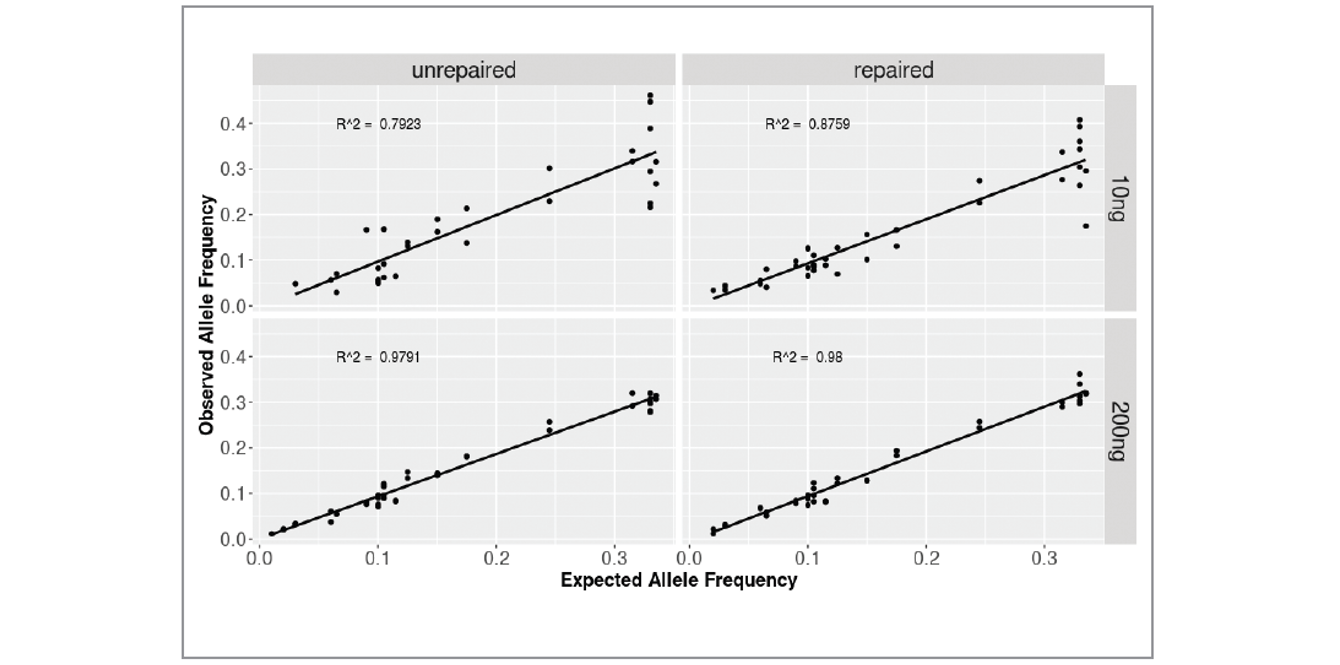
Figure 7: Correlation between expected and observed allele frequency in severely compromised fcDNA unrepaired/repaired samples at 10 and 200 ng input. The R2 values are improved in repaired samples, most noticeably at 10 ng, the lowest input tested.
†Kind gift from Horizon Discovery, 8100 Cambridge Research Park, Waterbeach, Cambridge CB25 9TL, United Kingdom
SureSeq: For Research Use Only; Not for Diagnostic Procedures.
Call +44 (0)1865 856800 Email contact@ogt.com
Send us a message and we will get back to you
 Visit International site
Visit International site Visit Canada site
Visit Canada site